
Author: Shelley Pallan
Institution: York University
Date: October 2006
ABSTRACT
Obesity has been linked to the pathogenesis of heart failure. Leptin, a 16-kD peptide hormone synthesized mainly by the adipocytes reduces food consumption and enhances energy expenditure. Leptin receptors (Ob-R) consist of six isoforms (ObRa-ObRf) and are present mainly in the hypothalamus as well as other tissues. Leptin deficiency and resistance have been implicated in the development of left ventricular hypertrophy and myocardial infarction. Impaired leptin action can also affect insulin sensitivity in cardiomyocytes leading to development of heart failure. The effects of leptin on cardiomyocytes have not been analysed in a comprehensive manner. This review explores how leptin-induced signaling pathways and leptin deficiency or resistance can promote heart failure by promoting cardiomyocyte hypertrophy, maladaptive vascularremodeling, impairing cardiac contractility, and perturbing fatty acid metabolism in cardiac myocytes.
INTRODUCTION
A. Obesity and heart failure
Obesity has become an epidemic in Western society (Kenchaiah et al. 2004). Several epidemiological studies have shown a direct relationship between obesity and cardiovascular morbidity and mortality (Lavie et al. 2003; Kalantar-Zadeh et al. 2004). The Framingham Heart Study indicates that an elevated body-mass index (BMI) is an independent risk factor for predicting heart failure (HF) (Kenchaiah et al. 2002). HF is a complex syndrome that involves endothelial abnormalities, left-ventricular dysfunction, myocardial infarction and maladaptive vascular remodeling (Kenchaiah S, Narula J et al. 2004).
Leptin, a 16-kDa peptide hormone encoded by the obesity (ob) gene (Sweeney 2002), is emerging as a novel mechanistic link between obesity and cardiovascular disease (Sader et al. 2003). It is mainly synthesized and secreted by adipocytes (Sweeney 2002), but also by other tissues such as the liver, kidney, lungs, pancreas and heart (Sader et al. 2003). Clinical studies have shown that serum leptin levels are a strong predictor of myocardial infarction in men and women as well as a predictor of future cardiovascular events in patients with atherosclerosis (Wolk et al. 2004). Leptin levels are also increased after myocardial infarction in hypertensive patients independently of traditional risk factors (Wallerstedt et al. 2004).
Basic science research into the effects of leptin on cardiomyocytes is still an emerging field. The objectives of this review are to discuss the intracellular signaling pathways by which leptin regulates cardiomyocyte hypertrophy, mitosis, contractility and myocardial extracellular matrix remodeling and fatty acid metabolism in order to elucidate the role leptin plays in the development and progression of heart failure. In addition, the pathway by which leptin deficiency perturbs insulin-mediated glucose metabolism will be explored.
B. Cardiomyocyte remodeling in response to vascular injury
Cardiomyocytes are muscle cells important in the synchronized contraction of the heart (Schwartz et al. 2001; Pennisi et al. 2002). Until recently, cardiomyocytes were considered terminally differentiated cells which undergo hypertrophy during postnatal maturation and adulthood and in conditions of increased workload (Anversa et al. 2002). However, numerous studies indicate that myocyte proliferation and hyperplasia can occur in physiological states and in pathological remodeling (Anversa et al. 2002). Cardiac remodeling is characterized by changes in gene expression and molecular signaling that result in abnormal cardiac function due to alterations in the size and shape of atria, ventricles, cardiac valves and blood vessels (Fedak et al. 2005). Cardiomyocyte proliferation is enhanced acutely after myocardial injury but is attenuated in end-stage cardiac failure (Anversa and Nadal-ginard. 2002). It is still not completely understood whether cardiomyocyte mitosis exerts salutary or adverse effects on ventricular remodeling (Leri et al. 2002). For instance, myocytes that have recently undergone mitosis cause cavitary dilation leading to thinning of the ventricular wall whereas myocyte proliferation involving the parallel addition of cells increases wall thickness and initially improves myocardial function (Leri et al. 2002). Cardiomyocyte apoptosis and necrosis have also been implicated in the structural deformities of the stressed myocardium (Anversa et al.. 2002). At the initial stages of myocardial injury, cardiac remodeling is an adaptive response that involves alterations in the size and number of cardiomyocytes (Lorell 1997). However, a sustained hypertrophic response entails maladaptive remodeling that develops into clinical pathologies such as left ventricular hypertrophy, myocardial infarction, and chronic heart failure (Lorell. 1997).
C. Leptin signaling in the heart
Physiologically, leptin reduces food intake and enhances energy expenditure when it binds to its receptors in the hypothalamus (Sader et al. 2003). Leptin also affects insulin-mediated glucose metabolism (Cohen et al. 1996; Liu et al. 1997; Emilsson et al. 1997). The leptin receptors are localized in the brain, pancreas, liver and heart (Sader et al. 2003). Leptin receptors are encoded by the diabetes (db) gene whose pre-mRNA transcript is alternatively spliced to produce six isoforms: ObRa, ObRb, ObRc, ObRd, ObRe and ObRf (Sweeney 2002). The rat heart expresses the receptor isoforms Ob-Ra, Ob-Rb and ObRe with a regional and gender-specific distribution (Purdham et al. 2004). Ventricular cardiomyocytes express the receptor isoform ObRa, which contains a shorter intracellular domain, in higher concentrations than the other isoforms (Purdham et al. 2004). Although the source of leptin in the coronary effluent has not been determined, leptin is detected in serum-free media containing cultured rat neonatal ventricular myocytes and a steady efflux of leptin has also been shown in isolated perfused rat hearts (Purdham et al. 2004). Thus, leptin may endogenously regulate cardiac function in an autocrine and paracrine manner.
Leptin regulates various signal transduction mechanisms such as the janus kinase/ signal transducer and activator of transcription (JAK/STAT), phosphatidylinositol-3-kinases (PI 3-kinase), and mitogen-activated protein kinase (MAPK) signaling pathways (Sweeney, 2002). Furthermore, leptin also affects cellular signaling in various cell types via Src-like homology-2 (SH2) domain in tyrosine phosphatases (SHP-2), suppressors of cytokine signaling (SOCS), insulin receptor substrate (IRS) proteins, protein kinase B, protein kinase C, cyclic AMP, nitric oxide, Rho GTPases, phospholipase C, voltage-gated Ca2+ channels and reactive oxygen species (ROS) (Sweeney 2002). To date, the specific functions of many of the leptin-regulated signal transducing pathways have not been elucidated in cardiomyocytes.
In the healthy state, plasma leptin levels correspond to the mass of adipose tissue (Sader et al. 2003). Leptin deficiency in ob/ob mice leads to increased food consumption, lowered energy expenditure and ventricular hypertrophy (Sader et al. 2003). In db/db mice, the leptin receptor gene has been ablated and the leptin receptor gene in fa/fa
rats contains a missense mutation in its extracellular domain that renders the receptor non-functional (Hiraoka-Yamamoto et al. 2004). The leptin-resistant state arising from leptin receptor insensitivity has been widely observed in clinically obese patients who also suffer insulin resistance (Sader et al. 2003). Plasma leptin levels are independent predictors of cardiovascular complications (Wallace et al. 2001), yet it still remains to be resolved whether leptin is a pro-hypertrophic or anti-hypertrophic hormone.
MECHANISMS OF ACTION OF LEPTIN ON CARDIOMYOCYTES
A. Effects of leptin on cardiomyocyte hypertrophy
Endothelin-1 (ET-1) is a vasoconstrictor secreted primarily by endothelial cells and also by macrophages, fibroblasts and cardiac myocytes in response to mechanical and chemical stimuli (Yamazaki et al. 1996). Endothelin-1 levels are elevated in patients with heart failure and ET-1 also augments protein synthesis and surface area of cardiomyocytes without cell proliferation (Yamazaki et al. 1996). In neonatal cardiomyocytes in vitro, the levels of endothelin-1 and intracellular reactive oxygen species (ROS) are significantly elevated in a dose-dependent manner upon leptin treatment as compared to the controls (Xu et al. 2004). Leptin-induced hypertrophy in cardiomyocytes manifests as a significant increase in total levels of RNA, atrial natriuretic peptide (ANP), cell surface area and (3H)leucine incorporation, a measurement of the rate of protein synthesis (Xu et al. 2004). These effects are abrogated upon treatment with endothelin-1-A (ETA) receptor antagonist, atrasentan (ABT-627) or catalase, a peroxide decomposing enzyme (Xu et al. 2004). ET-1 alone also significantly elevates hypertrophic responses which are inhibited by ABT-627 and catalase (Xu et al. 2004). However, it has not been shown whether the hypertrophic responses are more pronounced when the cardiomyocytes are pre-incubated with increasing concentrations of leptin only or endothelin-1 alone. Furthermore, ET-1 also induces ROS production in cardiomyocytes which is reduced upon pre-treatment with ABT-627 and the antioxidant, catalase (Xu et al. 2004). These results suggest that leptin-induced hypertrophy may cause chronic oxidative stress and inflammation in the myocardium, similar to other agents such as tumor necrosis factor-α (TNF-α), norepinephrine, and angiotensin II which also induce hypertrophy via ROS upregulation (Nakamura et al. 1998, Luo et al. 2001). Endothelin-1Areceptor blockade did not entirely abrogate leptin-induced ROS production (Xu et al. 2004) indicating that other mechanisms may also produce ROS and regulate cardiomyocyte hypertrophy Another study suggests that cardiomyocyte hypertrophy induced by endothelin-1 increased phosphorylation and activation of extracellular signal-regulated kinase-1/2 (ERK1/2) and involved upregulation of protein kinase C (PKC) and Raf-1 kinase (Yamazaki et al. 1999). However, it still remains to be examined whether leptin also upregulates PKC and Raf-1 kinase expression in cardiomyocytes. Ventricular cardiomyocytes isolated from mice with a point mutation in the ob gene exhibit impaired contractile function which is significantly ameliorated by local leptin administration. Interestingly, rats treated with bostenan, an endothelin-1 receptor antagonist showed a significant reduction in myocardial infarction mortality (Ostrowski et al. 2003). Hence, the in-vitro and animal studies suggest that endothelin receptor antagonists and agents that downregulate ROS may be clinically administered to alleviate hypertrophy and therby thwart the progression of heart failure. Simvastatin, a cholesterol-lowering drug decreases leptin-induced ROS-mediated hypertrophy in rat neonatal cardiomyocytes (Hu et al. 2006).
In clinical settings, left ventricular hypertrophy (LVH) is observed in patients with elevated leptin levels (Sader et al. 2003). However, some studies provide paradoxical evidence which suggests that leptin may act as ant-hypertrophic hormone. An intriguing study by Barouch et al. shows that leptin deficiency leads to ventricular hypertrophy in ob/ob and db/db mice but not in wild-type controls (Barouch et al. 2003). In ob/ob mice, weight loss was induced either by leptin infusion or by caloric restriction (Barouch et al. 2003). Histologically, leptin infusion reduced cardiomyocyte hypertrophy in ob/ob mice to a much greater degree than caloric restriction and it also reversed left ventricular hypertrophy to almost normal states in ob/obmice, whereas caloric restriction did not significantly reduce ventricular hypertrophy (Barouch et al. 2003). These results implicate that leptin and its downstream signaling mediators have an anti-hypertrophic effect, independently of body mass when leptin's signaling mechanism is functional. Furthermore, hemodynamic changes in ob/ob mice did not contribute significantly to increased LV mass because parameters such as heart rate, systolic blood pressure, and LV end-diastolic volume did not differ between ob/ob and ob/+ mice prior to leptin infusion (Barouch et al. 2003). Hence, this study implies that leptin maintains normal cardiomyocyte morphology and left ventricular wall thickness independently of hemodynamic changes. In addition, clinical studies by Tritos et al. (1999) show that normotensive obese men have a higher absolute and height-indexed left-ventricular mass than lean men and normotensive obese men also exhibit a positive relationship between serum leptin levels and height-indexed left ventricular mass (because they are leptin-resistant) but this association is not observed after adjustment for body mass index (Tritos et al. 1999). The Tritos et al. clinical trials (1999) support the Barouch et al. observations that hemodynamic load is not the most pivotal factor in the pathogenesis of left-ventricular hypertrophy in obesity. But the following discussion of selective leptin resistance' provides contradictory evidence.
Selective leptin resistance
A relatively new concept of selective leptin resistance' which is observed in certain mouse models such as the agouti, yellow obese mice and mice with diet-induced obesity, holds that partial resistance to the effects of leptin on thermogenic metabolism and satiating appetite can occur in some states of obesity (Mark et al. 2004). However, the sympathetic excitatory effects of leptin such as increased arterial pressure and renal sympathetic nerve activity are preserved in hyperleptinemia (Mark et al. 2004). This concept emerged from studies that showed lowered arterial pressure in grossly obese ob/ob mice as compared to lean, wild-type controls and leptin infusion in ob/ob mice increased arterial pressure in spite of an accompanying loss in body weight (Mark et al. 2004). Hence, the hemodynamic actions of leptin in a state of hyperleptinemia are mediated mainly by sympathetic activation. The concept of selective leptin resistance challenges the Barouch et al. (2003) conclusion which states that leptin infusion exerts a direct anti-hypertrophic effect independently of hemodynamic load based on results that showed similar hemodynamic parameters in ob/oband ob/+ mice prior to leptin reconstitution. Leptin infusion in ob/ob mice would alter hemodynamic parameters due to the sympathetic excitatory effect of the hormone (in the presence of functional leptin receptors); this factor could also be contributing to decreased LV wall thickness and LV mass that Barouch et al. observed.
B. Effects of leptin on cardiomyocyte mitosis
Mitogen-activated protein kinases (MAPK) such as ERK1/2 and p38 are involved in intracellular signal transduction pathways that regulate cell proliferation and differentiation (Tajmir et al. 2004). In the Tajmir et al. study (2004). the effect of leptin on MAP kinases was studied in a stable murine cardiomyocyte cell line (HL-1 cells). The approximate plasma leptin concentration in obese individuals, 6nM, caused a significant time- and dose-dependent increase in cell number (Tajmir et al. 2004). Leptin induced the proliferative response through the activation of ERK1/2 and PI-3-kinase signaling pathways as indicated by elevated levels of phosphorylated ERK1/2 and the association of p85 regulatory subunit of PI-3-kinase with phosphotyrosine immunoprecipitates (Tajmir et al. 2004). Bromodeoxyuridine incorporation assays also showed enhanced DNA replication in murine and human pediatric cardiomyocytes upon leptin treatment (Tajmir et al. 2004).
The Karmazyn group has shown that leptin acutely increased the levels of phospho-ERK1/2 and phospho-p38 MAP kinase in rat neonatal cardiomyocytes after 5 and 10 minutes of leptin treatment, respectively (Rajapurohitam et al. 2003). The leptin-induced p38 activation is sustained for a longer period of time as compared to ERK1/2 activation, which suggests that p38 signaling and its downstream transcription factors may be involved in the long-term maladaptive cardiac remodeling in obese patients suffering from chronic heart failure (Rajapurohitam et al. 2003). A p38 inhibitor, SB203580, attenuated the leptin-induced hypertrophic responses as shown by decreased (3H) leucine incorporation and attenuated the expression of α-skeletal actin and myosin light chain-2 (MLC-2) (Rajapurohitam et al. 2003). The p38 inhibitor did not enhance apoptosis at concentrations of leptin up to 3.1nM (Rajapurohitam et al. 2003); these findings have some implications in determining the threshold plasma leptin concentration that triggers cardiac apoptosis. However, the p38 isoform(s) through which leptin mediates its effects remain to be elucidated. Interestingly, the ERK1/2 inhibitor, PD98059, did not significantly alter the hypertrophic responses such as cell surface area, α-skeletal actin mRNA, MLC-2 mRNA, and (3H) leucine incorporation in cardiomyocytes pre-treated with leptin (Rajapurohitam et al. 2003). The insignificant change in (3H) leucine incorporation is intriguing because it has been shown that PD98059 significantly attenuates the leptin-induced proliferative activity and DNA replication in murine and pediatric cardiomyocytes (Tajmir et al. 2004). Hence, (^3H) leucine incorporation in cardiomyocytes treated with leptin and PD98059, is also expected to decrease concomitantly due to lowered amounts of proteins involved in regulation of cell growth. However, the different cell lines used in the two studies may underlie the discrepancies. Leptin in this case may be beneficial in promoting mitosis in cardiomyocytes. However, it remains to be determined which dose of leptin would be optimal in promoting myocyte apoptosis without initiating maladaptive hypertrophy as one of the causes of heart failure is cardiomyocyte apoptosis and necrosis (Williams SD et al. 2006). Hence, leptin can have beneficial or adverse effects in the progression of heart failure depending on the cellular response it induces.
C. Effects of leptin on cardiomyocyte contractility
Impaired myocardial contractility is observed in obese individuals (Anversa and Nadal-ginard 2002) and serum levels of nitric oxide (NO), a vasodilator, are increased following leptin administration (Fruhbeck et al. 1999). Nickola et al. (2000) have assessed the effects of leptin on cardiomyocyte contraction by assessing parameters such as cell shortening, an indicator of the contractile response, intracellular Ca2+ transients ((Ca2+)i) and nitric oxide synthase (NOS) activity in rat ventricular myocytes (Nickola et al. 2000). Leptin inhibited myocyte shortening (22.4% maximally) in a concentration-dependent manner; it was shown that leptin-induced attenuation of cardiomyocyte shortening could occur due to changes in intracellular concentrations of Ca2+ (Nickola et al. 2000). Fura fluorescence intensity (FFI) staining following the treatment of myocytes with fura-2-AM, a cell permeable fluorescent Ca2+indicator, showed that leptin reduced free intracellular Ca2+ (24.2% maximally) (Nickola et al, 2000). A recent study has shown that nitration of sarcoplasmic reticulum Ca2+-ATPase (SERCA2a isoform) due to increased production of peroxynitrites in the hearts of patients with idiopathic dilated cardiomyopathy without coronary artery disease can lead to impaired myocardial function (Lokuta et al. 2005). But, the specific mechanism by which leptin reduces intracellular calcium concentrations, such as its effects on sarcoplasmic reticulum Ca2+-ATPase pump or the plasma membrane Na+-Ca2+ exchanger have not been investigated. A potential mechanistic link between leptin action and nitric oxide on cardiomyocytes was elucidated using Nω-nitro-L-arginine-methyl ester (L-NAME), a selective NOS inhibitor (Nickola et al. 2000). Indeed, leptin-induced reduction in both cardiomyocyte shortening and intracellular Ca2+ transients was blunted in the presence of L-NAME (Nickola et al. 2000). In addition, leptin enhanced nitric oxide synthase (NOS) activity in a dose-and time-dependent manner (Nickola et al. 2000). The specific isoforms of constitutive NOS that mediate leptin's effects have not been elucidated. Hence, leptin-induced nitric oxide production may lead to impaired cardiomyocyte contractility and elevated levels of serum nitric oxide can contribute to cardiac remodeling leading to heart failure. Enhanced nitric oxide production may also increase the ROS levels contributing to impaired contractility as well as maladaptive hypertrophy.
Leptin-induced cardiac responses are mediated partially via the JAK/STAT signaling pathway (Wold et al. 2002). In isolated rat ventricular myocytes from spontaneously hypertensive rats (SHR), leptin-induced depression of myocyte shortening and intracellular Ca2+ transients was mitigated as compared to the normotensive control mice (Wold et al. 2002). This effect is similar to the blunted leptin-induced myocyte shortening that is observed upon administration of L-NAME (Nickola et al, 2000). Leptin-induced enhancement in NOS activity was also blunted in hypertensive rat ventricular myocytes even though the basal levels of NO were comparable in SHR and control groups (Wold et al. 2002). Furthermore, elevated extracellular calcium levels did not significantly affect the attenuated response to leptin-induced inhibition of myocyte contractility in hypertensive mice and varying concentrations of leptin also did not affect intracellular Ca2+ transients (Wold et al. 2002). This indicates that intracellular signaling mechanisms, instead of calcium concentrations, are responsible for the cardioprotective effect of hypertension. In normotensive rats, the leptin-induced inhibition of myocyte contraction was prevented by L-NAME, AG-490, a JAK-2 inhibitor and SB203580, a p38 inhibitor (Wold et al. 2002). This implies that leptin exerts its adverse effect on contractility through the NO, MAP kinase and JAK/STAT pathways (Wold et al. 2002). Interestingly, in spontaneously hypertensive myocytes, the leptin-induced inhibition of myocyte shortening was actually augmented by treatment with AG-490 and SB203580 (Wold et al. 2002). Thus, the state of hypertension alters the signal transduction of MAP kinase and JAK/STAT pathways (by a yet unknown mechanism) to attenuate leptin-induced cardiomyocyte dysfunction. But when these pathways are inhibited, hypertension cannot protect the myocytes from the leptin-induced reduction in contractile response. Such an intricate hypothesis is supported by evidence indicating that even though leptin enhanced phosphorylation of STAT3 in cardiomyocytes in both hypertensive and normotensive rats as compared to the controls (not treated with leptin), the hypertensive myocytes showed substantially elevated basal STAT3 phosphorylation levels (without leptin treatment) (Wold et al. 2002). The comparable levels of Ob-R receptors in both hypertensive and normotensive rats support the hypotheses that perturbations in intracellular leptin signaling instead of receptor downregulation lead to the divergent responses upon leptin exposure. However, these studies have not anaylsed NO's role in promoting hypertrophy and impairing contractility concomitantly. As such, it remains to be elucidated whether the impairment in contractile function of cardiomyocytes is independent of the hypertrophic response or a result of hypertrophy.
D Effects of leptin on extracellular matrix remodeling in the myocardium
Sustained vascular injury may promote maladaptive matrix remodeling by de-regulating collagen production (Sundstrom et al. 2006). The turnover rate for type I and type III collagen is a predictor of cardiovascular complications (Sundstrom et al. 2006). One study shows that leptin increases the expression of matrix metalloproteinase-2 (MMP-2), increases collagen type III and IV mRNA and decreases collagen type I mRNA, but does not affect total collagen synthesis in HL-1 cardiomyocytes (Lee et al. 2006). This suggests that leptin selectively regulates different forms of collagen although further studies are required to corroborate the regulation of collagen synthesis by leptin and further explore the effects of leptin on other components of the matrix such as proteoglycans and adhesive proteins.
The effects of leptin on cardiomyocytes have only begun to be elucidated. In pathological conditions, perturbations of leptin signaling and other signal transduction pathways regulated by leptin in cardiomyocytes underlie the pathology of cardiomyocyte hypertrophy. In particular, alterations in nitric oxide, JAK/STAT, MAP kinase, and beta-adrenergic pathways have been implicated in the hypertrophic response. Further in vitro studies investigating the integrated effects of leptin on cardiomyocytes via signaling pathways of SOCS-3, Protein kinase B, Rho GTPases and other signaling pathways regulated by the hormone (Sweeney 2002) could provide a more comprehensive understanding of leptin action on cardiac myocytes.
E. Effects of leptin deficiency on fatty acid metabolism in cardiomyocytes
During excess caloric intake, leptin regulates lipid homeostasis by preventing accumulation of triglycerides in non-adipose tissues, such as the myocardium, which are not specialized to store fatty acids (Unger. 2002). Leptin deficiency or leptin resistance perturb the liporegulation and result in lipotoxicity, lipoapoptosis and generalized steatosis, the process of tissue degeneration characterized by deposition of fat globules in cells (Unger 2002). Ultimately, lipotoxicity results in pathologic conditions such as cardiomyopathy which is an aspect of the metabolic syndrome (Unger 2002). Recent studies have implicated that leptin is an anti-steatotic hormone (Unger 2002). A high-fat diet in control rodents caused a 150-fold increase in body fat, but the myocardium exhibited only a minimal rise in triglyceride content because of the state of hyperleptinemia in obesity (Unger 2002). However, in ob/ob, db/db and fa/faanimals, non-adipose tissues such as the cardiomyocytes exhibited significantly higher triglyceride contents as compared to wild-type obese controls (Unger 2002). In obese Zucker diabetic fatty (ZDF) fa/fa rats, echocardiographic examination showed impaired myocardial contractility that occurred due to cardiomyocyte apoptosis (Zhou et al. 2000).
Two mechanisms for intracellular accumulation of lipids in non-adipose tissues have been proposed: increased de novo lipogenesis and decreased compensatory beta-oxidation of fatty acids (Unger 2002). In the cardiac muscle of 14-week old fa/fa leptin-resistant rats, biochemical analyses showed enhanced mRNA expression of genes encoding for the enzyme glycerol phosphate acyl transferase (GPAT) which is involved in fatty acid esterification (Zhou et al. 2000). At the initial stages of fatty acid excess, triglyceride formation prevents free fatty acids from entering deleterious metabolic pathways (Unger 2002). Concomitantly, the expression of enzymes involved in beta-oxidation of fatty acids such as acyl-CoA oxidase (ACO) and carnitine palmitoyltransferase (CPT-1), which also regulates mitochondrial uptake of fatty acids and their transcription factor, peroxisome proliferator-activated receptor-α (PPAR-α) were attenuated in the fa/fa rats (Zhou et al. 2000). However, a strikingly different study has shown enhanced expression of enzymes implicated in beta-oxidation such as long-chain acyl-CoA dehydrogenase (LCAD), diacylglycerol acyltransferase (DGAT) and CPT-1 in 10- to 11-week old ob/ob mice (Christoffersen et al. 2003). This discrepancy may be due to different species of animals and the different types of genes deleted in the latter two studies.
In the Zhou et al. study, the aberrant expression of CPT-1 and ACO was accompanied by a rise in ceramide, an apoptotic mediator, which activated the expression of inducible nitric oxide synthase (iNOS) in the myocardium of fa/fa rats (Zhou et al. 2000). A resulting increase in nitric oxide (NO) and peroxynitrites also accelerates apoptosis. The hydrolysis of the increased stores of triglycerides to fatty acyl-CoA provides elevated substrate for ceramide synthesis (Zhou et al. 2000). Ceramide also activates various isoforms of protein kinase C (PKC), a serine/threonine kinase, which can induce transcription of specific genes via MAP kinase and NF-χB signaling mechanisms (Ruvolo et al. 2003; Murphy et al. 2005). Elevated levels of PKC isoforms, especially PKC ß, are implicated in the development of cardiomyocyte hypertrophy and transition to heart failure (Murphy et al. 2005).
Another study showed an increased expression of genes controlling fatty acid metabolism in ob/ob mouse hearts (Christoffersen et al. 2003). The following genes were significantly upregulated in mouse ob/ob hearts as compared to the ob/+ hearts: lipoprotein lipase which generates free fatty acids, genes encoding proteins implicated in fatty acid transport across myocyte cell membrane such as CD36, fatty acid transport protein (FATP)-1 and FATP-4 and intracellular fatty acid transport such as heart-specific fatty acid binding protein (hFABP) (Christoffersen et al. 2003). The enhanced levels of these enzymes promote accumulation of free fatty acids in cardiomyocytes which can then enter non-oxidative metabolic pathways that produce pro-apoptotic molecules such as ceramide, iNOS and NO (Christoffersen et al. 2003). Indeed, elevated concentration of NO is a pivotal factor in promoting heart failure as it is involved in maladaptive hypertrophy, impaired contractility and myocyte apoptosis.
Prolonged leptin deficiency or leptin resistance leads to uncompensated caloric excess in cardiomyocytes (Unger. 2002). In the absence of leptin action, the expression of PPAR-α is reduced and PPAR-γ is elevated; the excess fatty acids bind to PPAR-γ which activates transcription of lipogenic enzymes such as acetyl-CoA carboxylase (ACC) and fatty acid synthetase (FAS) (Unger, 2002). Treatment with the anti-steatotic agent troglitazone (TGZ) reduces cardiac ceramides and triglycerides and enhances myocardial contractility (Zhou et al. 2000). Inhibitors of iNOS and ceramide synthesis are also potential therapeutic anti-steatotic agents. In humans, initial stages of diet-induced obesity are not accompanied by cardiovascular complications, suggesting that leptin may confer anti-steatotic protection (Unger 2002). At the later stages of human obesity, it remains to be fully elucidated whether lipotoxicity or leptin resistance leads to adverse cardiovascular events.
Furthermore, increased accumulation of lipids in
ob/ob mice cardiomyocytes leads to cardiac dysfunction. Histological staining of obese ob/obmice ventricular cardiomyocytes exhibited a marked accumulation of neutral lipids (a 3-fold increase in triglyceride contents) as compared to non-diabetic lean ob/+ controls (Christoffersen et al. 2003).
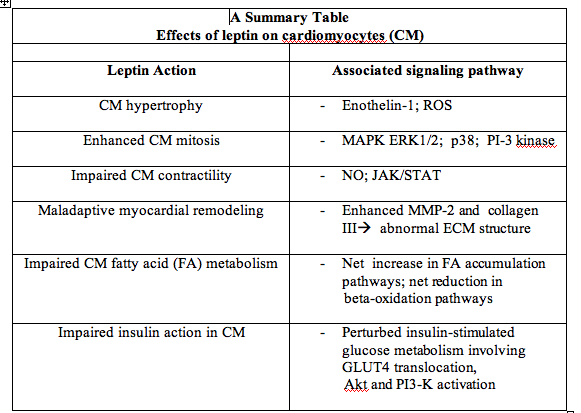
article_808_order_0
Acyl-CoA synthase (ACS) is an enzyme that activates and esterifies fatty acids (Lee et al, 2004). More evidence of leptin's anti-steatotic function arises from a study showing that hyperleptinemia attenuated lipotoxic cardiomyopathy in ACS-transgenic mice (Lee et al, 2004). Hyperleptinemia was induced by injection of recombinant adenovirus containing the leptin cDNA in transgenic mice with severe cardiomyopathy that was caused by cardiomyocyte-specific overexpression of mice acyl-CoA synthase; leptin expression restored normal triglyceride contents in mice cardiomyocytes (Lee et al. 2004). However, normoleptinemic control ACS-transgenic mice developed cardiac hypertrophy as manifested by thickened ventricular walls and histological disorganization of myofibers and interstitial fibrosis with cytoplasmic lipid vacuoles (Lee et al, 2004). This study corroborates previous experiments showing that leptin lowers intracellular triglyceride pools. In the hyperleptinemic ACS-transgenic mice, signaling mechanisms involving protein kinase phosphorylation could have enhanced beta-oxidation in cardiomyocytes which resulted in lowered lipid vacuoles. Hence, it is possible that the hyperleptinemia in obesity protects cardiomyocytes from lipotoxicity. The combined effects of triglycerides stores and free fatty acids entering non-oxidative pathways in a leptin-deficient state ultimately lead to lipoapoptosis in cardiomyocytes. Clinically, obesity is currently associated with coronary artery diseases but recent studies suggest that lipotoxic heart disease can also contribute to impaired myocardial function.
Taken together, these studies suggest that leptin is a central regulator of cardiac fatty acid metabolism and leptin signaling channels lipids into oxidative pathways and prevents accumulation of lipids in cardiomyoyctes. Lipotoxicity of cardiomyocytes is likely a key mechanism in the pathogenesis of heart failure in obesity because the products of nonoxidative metabolic pathways such as iNOS could synergistically interact with disrupted leptin signaling and result in myocyte apoptosis and cardiovascular complications. The generation of murine models or chimeric mice models that have ablated genes encoding for a signaling molecule(s) regulated by leptin can also yield insightful results in hypo- or- hyperleptinemic states. The discovery of leptin-receptor agonists and antagonists and leptin-inhibition in cardiomyocytes of healthy animals will shed more light on the physiological role of the hormone.
EFFECTS OF LEPTIN ON INSULIN SIGNALING IN CARDIOMYOCYTES
F. Cross talk between leptin and insulin signaling.
Hyperleptinemia has been implicated in impaired insulin sensitivity (Leyva et al. 1998). Insulin also stimulates the expression of the ob gene and a positive relationship between serum leptin levels and lowered insulin sensitivity is found in patients with congestive heart failure after correcting for total body fat (Leyva et al. 1998). Research into the interrelationship between leptin and insulin signaling specifically in cardiomyocytes will be useful in understanding insulin-resistance and cardiovascular complications that accompany diabetes in obese patients Not surprisingly, signaling pathways of leptin and insulin exhibit cross-talk; both ob/ob and db/db mice are hyperglycemic and hyperinsulinemic (Leyva et al. 1998). However, very few studies have investigated the relationships between leptin and insulin signaling in cardiomyocytes and in chronic heart failure.
Recently, Mazumder et al (2004) showed that altered insulin action in ob/ob mice was responsible for impaired insulin-stimulated glucose uptake and substrate metabolism by assaying various components of insulin signaling pathways. The phosphorylation levels of Akt and the p85 subunit of PI-3-kinase, two components in insulin signaling were similar in ob/obmice and wild-types (Mazumder et al. 2004). Upon insulin stimulation, IR phosphorylation and activation of Akt and PI-3-kinase were significantly lower in ob/ob mice cardiomyocytes (Mazumder et al. 2004). Even though basal rates of glucose uptake are similar in ob/ob and wild-types, insulin-stimulated glucose uptake was completely blunted in ob/ob cardiomyocytes (Mazumder et al. 2004). The decline in glucose uptake was not associated with decreased total GLUT4 expression, which suggests that GLUT4 translocation could be impaired due to perturbed insulin signaling (Mazumder et al. 2004).
In addition, leptin deficiency also affects insulin-mediated substrate metabolism (Mazumder et al. 2004, Kong et al. 2002). The rates of glycolysis (under low palmitate concentration) were 22% lower in ob/ob hearts and insulin stimulation enhanced glycolysis in wild-types but not ob/ob mice (Mazumder et al. 2004). This suggests that leptin can also promote glucose metabolism when long-chain fatty acids such as palmitate are present in low amounts. However, the signaling pathways by which leptin promotes insulin-mediated glucose metabolism have not been explored. It may be that leptin lowers fatty acid pools in cardiomyocytes to promote glucose oxidation.
Furthermore, ob/ob hearts have a higher rate of fatty acid utilization regardless of presence or absence of insulin (Kong et al. 2002). Insulin also reduces palmitate levels which are implicated in cardiomyocyte apoptosis in wild-type mice but not in ob/ob mice (Kong et al. 2002). Hence, leptin deficiency leads to impaired insulin-stimulated glucose metabolism; these studies provide clues to the mechanisms by which leptin regulates downstream mediators in insulin signaling.
G. Effects of PPAR-γ agonists on cardiomyocytes and cardiac function in leptin resistant (db/db) mice
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors belonging to the nuclear receptor family and are implicated in whole-body insulin sensitivity (Carley et al. 2004). There are three known isoforms of the receptor, PPAR-α, PPAR-ß/γ and PPAR-γ (Carley et al. 2004). The alpha isoform is highly expressed in the heart and it regulates fatty acid uptake and oxidative metabolism (Carley et al, 2004). As compared to PPAR-α , PPAR-γ expression in cardiomyocytes is very low and it was thought that PPAR-γ exerted no direct effects on fatty acid and glucose metabolism in cultured cardiomyocytes (Carley et al. 2004). But recently, Carley et al. (2004) have shown that in diabetic db/db mice cardiomyocytes, treatment with a novel PPAR-γ agonist, 2-(2-(4-phenoxy-2-propylphenoxy)ethyl)indole-5-acetic acid (COOH) restored blood glucose to normal levels and insulin-stimulated glucose uptake was also enhanced (Carley et al. 2004). The COOH-treated cardiac myocytes also exhibited enhanced glucose oxidation and reduced palmitate oxidation (Carley et al. 2004). However, the COOH agonist did not ameliorate cardiac contractility (Carley et al. 2004). This study provides an indirect link between leptin and PPARγ-induced transcriptional activation of genes that can potentially lead to insulin sensitization since an imbalance in the relative concentrations of PPARs is present in the leptin-resistant db/db mice.
Considering the current gaps in knowledge regarding the effects of leptin on cardiomyocytes, this emerging field is a bright avenue for future research.
ACKNOWLEDGEMENTS.
I am very grateful to Dr. Gary Sweeney at York University.
REFERENCES
Anversa P, Nadal-Ginard B. (2002). Myocyte renewal and ventricular remodelling. Nature. 415: 240-3.
Anversa P, Leri A, Kajstura J, Nadal-Ginard B. (2002). Myocyte growth and cardiac repair. J Mol Cell Cardiol. 34: 91-105.
Barouch LA, Berkowitz DE, Harrison RW, O'Donnell CP, Hare JM. (2003). Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation. 108: 754-9.
Carley AN, Semeniuk LM, Shimoni Y, Aasum E, Larsen TS, Berger JP, Severson DL. (2004). Treatment of type 2 diabetic db/db mice with a novel PPARgamma agonist improves cardiac metabolism but not contractile function. Am J Physiol Endocrinol Metab. 286: E449-55.
Christoffersen C, Bollano E, Lindegaard ML, Bartels ED, Goetze JP, Andersen CB, Nielsen LB. (2003). Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology. 144: 3483-90.
Cohen B, Novick D, Rubinstein M. (1996). Modulation of insulin activities by leptin. Science. 274: 1185-8
Correia ML, Haynes WG. (2004). Leptin, obesity and cardiovascular disease. Curr Opin Nephrol Hypertens. 13: 215-23.
Emilsson V, Liu YL, Cawthorne MA, Morton NM, Davenport M. (1997). Expression of the functional leptin receptor mRNA in pancreatic islets and direct inhibitory action of leptin on insulin secretion. Diabetes. 46: 313-6.
Fedak PW, Verma S, Weisel RD, Li RK. (2005). Cardiac remodeling and failure From molecules to man (Part I). Cardiovasc Pathol. 14(1):1-11.
Fruhbeck G. (1999). Pivotal role of nitric oxide in the control of blood pressure after leptin administration. Diabetes. 48: 903-8
Hiraoka-Yamamoto J, Nara Y, Yasui N, Onobayashi Y, Tsuchikura S, Ikeda K (2004). Establishment of a new animal model of metabolic syndrome: SHRSP fatty (fa/fa) rats. Clinical and Experimental Pharmacology and Physiology. 31: 107-109.
Kalantar-Zadeh K, Block G, Horwich T, Fonarow GC. (2004). Reverse epidemiology of conventional cardiovascular risk factors in patients with chronic heart failure. J Am Coll Cardiol 43: 14391444
Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, Kannel WB, Vasan RS. (2002). Obesity and the risk of heart failure. N Engl J Med. 347: 305-13.
Kenchaiah S, Gaziano JM, Vasan RS. (2004). Impact of obesity on the risk of heart failure and survival after the onset of heart failure. Med Clin North Am. 88: 1273-94
Kenchaiah S, Narula J, Vasan RS. (2004). Risk factors for heart failure. Med Clin North Am. 88: 1145-72.
Kong YJ, Rabkin SW. (2002). Palmitate-induced cardiac apoptosis is mediated through CPT-1 but not influenced by glucose and insulin. Am J Physiol Heart Circ Physiol. 282: H717 - 725.
Lavie CJ, Milani RV. (2003). Obesity and cardiovascular disease: the hippocrates paradox? J Am Coll Cardiol. 42: 677-9.
Lee MP, Sweeney G. (2006). Insulin increases gelatinase activity in rat glomerular mesangial cells via ERK- and PI-3 kinase-dependent signalling. : Diabetes Obes Metab. 8: 281-8.
Lee Y et al. (2004). Hyperleptinemia prevents lipotoxic cardiomyopathy in acyl CoA synthase transgenic mice. Proc Natl Acad Sci U S A. 101: 13624-9
Leri A, Kajstura J, Anversa P. (2002). Myocyte proliferation and ventricular remodeling. J Card Fail. 8: S518-25.
Leyva F, Anker SD, Egerer K, Stevenson JC, Kox WJ, Coats AJ. (1998). Hyperleptinaemia in chronic heart failure. Relationships with insulin. Eur Heart J. 19: 1547-51.
Liu YL, Emilsson V, Cawthorne MA. (1997). Leptin inhibits glycogen synthesis in the isolated soleus muscle of obese (ob/ob) mice. FEBS Lett. 411: 351-5.
Lokuta AJ, Maertz NA, Meethal SV, Potter KT, Kamp TJ, Valdivia HH, Haworth RA. (2005) Increased Nitration of Sarcoplasmic Reticulum Ca2+-ATPase in Human Heart Failure. Circulation.111: 988-995
Lorell BH. (1997). Transition from hypertrophy to failure. Circulation. 96:3824-3827
Luo JD, Xie F, Zhang WW, Ma XD, Guan JX, Chen X.. (2001). Simvastatin inhibits noradrenaline-induced hypertrophy of cultured neonatal rat cardiomyocytes. Br J Pharmacol. 132: 159-64
Mazumder PK, O'Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. (2004). Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 53: 2366-74.
Nakamura K, Fushimi K, Kouchi H, Mihara K, Miyazaki M, Ohe T, Namba M. (1998). Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circulation. 98: 794-9.
Nickola MW, Wold LE, Colligan PB, Wang GJ, Samson WK, Ren J. (2000). Leptin attenuates cardiac contraction in rat ventricular myocytes. Role of NO. Hypertension. 36: 501-5.
Nielsen LB, Bartels ED, Bollano E. (2002). Overexpression of apolipoprotein B in the heart impedes cardiac triglyceride accumulation and development of cardiac dysfunction in diabetic mice. J Biol Chem. 277: 27014-27020.
Ostrowski RP, Januszewski S, Kowalska Z, Kapuscinski A. (2003). Effect of endothelin receptor antagonist bosentan on plasma leptin concentration in acute myocardial infarction in rats. Pathophysiology. 9:249-256.
Pennisi DJ, Rentschler S, Gourdie RG, Fishman GI, Mikawa T. (2002). Induction and patterning of the cardiac conduction system. Int J Dev Biol. 46: 765-75.
Port JD, Bristow MR. (2001). Altered beta-adrenergic receptor gene regulation and signaling in chronic heart failure. J Mol Cell Cardiol. 33: 887-905
Purdham DM, Zou MX, Rajapurohitam V, Karmazyn M. (2004). Rat heart is a site of leptin production and action. Am J Physiol Heart Circ Physiol. 287: H2877-84
Rahmouni K, Haynes WG. (2004). Leptin and the Cardiovascular System. Recent Progress in Hormone Research. 59: 225-244
Rajapurohitam V, Gan XT, Kirshenbaum LA, Karmazyn M. (2003). The Obesity-Associated Peptide Leptin Induces Hypertrophy in Neonatal Rat Ventricular Myocytes.Circ Res.93: 277-9.
Sader S, Nian M, Liu P. (2003). Leptin: a novel link between obesity, diabetes, cardiovascular risk, and ventricular hypertrophy. Circulation. 108: 644-6.
Schwartz SM, Duffy JY, Pearl JM, Nelson DP. (2001). Cellular and molecular aspects of myocardial dysfunction. Crit Care Med. 10: S214-9
Sundstrom J, Vasan RS. (2006). Circulating biomarkers of extracellular matrix remodeling and risk of atherosclerotic events. Curr Opin Lipidol. 17: 45-53
Sweeney G. (2002). Leptin signaling. Cellular Signalling. 14: 655-663
Tajmir P, Ceddia RB, Li RK, Coe IR, Sweeney G. (2004). Leptin increases cardiomyocyte hyperplasia via extracellular signal-regulated kinase- and phosphatidylinositol 3-kinase-dependent signaling pathways. Endocrinology. 145:1550-5
Tritos NA, Kissinger KV, Manning WJ, Danias PG (2004). Association between ghrelin and cardiovascular indexes in healthy obese and lean men. Clin Endocrinol. 60: 60-6.
Unger RH. (2002). Lipotoxic diseases. Annu Rev Med. 53:319-36
Wallace AM, McMahon AD, Packard CJ, Kelly A, Shepherd J, Gaw A, Sattar N. (2001). Plasma leptin and the risk of cardiovascular disease in the west of Scotland coronary prevention study (WOSCOPS). Circulation. 104: 3052-6.
Wallerstedt SM, Eriksson AL, Niklason A, Ohlsson C, Hedner T. (2004). Serum leptin and myocardial infarction in hypertension. Blood Press. 13: 243-6.
Wang W, Zhu W, Wang S, Yang D, Crow MT, Xiao RP, Cheng H. (2004). Sustained beta1-adrenergic stimulation modulates cardiac contractility by Ca2+/calmodulin kinase signaling pathway. Circ Res. 95: 798-806
Williams SD, Zhu H, Zhang L, Bernstein HS. (2006). Adenoviral delivery of human CDC5 promotes G2/M progression and cell division in neonatal ventricular cardiomyocytes. Gene Ther. 13: 837-43
Wold LE, Relling DP, Duan J, Norby FL, Ren J. (2002). Abrogated leptin-induced cardiac contractile response in ventricular myocytes under spontaneous hypertension: role of JAK/STAT pathway. Hypertension. 39: 69-74
Yamazaki T, Komuro I, Kudoh S, Zou Y, Shiojima I, Hiroi Y, Mizuno T, Maemura K,
Kurihara H, Aikawa R, Takano H, Yazaki Y. (1996). Endothelin-1 is involved in mechanical stress-induced cardiomyocyte hypertrophy. J Biol Chem. 271: 3221-8.
Yamazaki T, Komuro I, Zou Y, Yazaki Y. (1999). Hypertrophic responses of cardiomyocytes induced by endothelin-1 through the protein kinase C-dependent but Src and Ras-independent pathways. Hypertens Res. 22:113-9
Xu FP, Chen MS, Wang YZ, Yi Q, Lin SB, Chen AF, Luo JD. (2004) Leptin induces hypertrophy via endothelin-1-reactive oxygen species pathway in cultured neonatal rat cardiomyocytes. Circulation. 110: 1269-75
Wolk R, Berger P, Lennon RJ, Brilakis ES, Johnson BD, Somers VK. (2004). Plasma leptin and prognosis in patients with established coronary atherosclerosis. J Am Coll Cardiol. 44:1819-24.
Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH. (2000). Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A. 97: 1784-9.40.