

KWING YEUNG CHAN, YAT HEI HUGO KWONG, DEI MEN SZETO
ABSTRACT
Green fluorescent protein (GFP), a fluorescent marker extracted from Aequorea victoria, has been a prominent tool for protein visualisation in modern biomedical research. When properly folded, it emits green fluorescence upon UV illumination. Furthering our understanding of GFP’s structure, maturation, and spectrochemical properties would allow for increased optimisation, variant development, and protein research applications. Additionally, understanding protein localisations and protein-protein interactions can provide insights into the functions of the proteome. Examples such as global analysis of protein locations and specific localisation of virulence factors using GFP have been outlined. Utilisation of split-GFP further enables the detection of protein-protein interactions with high accuracy. Applications of split-GFP for probing protease activity and protein quantification in the context of neurodegenerative diseases have been further showcased. All these demonstrations manifest the potential of GFP in future proteomic studies, hence providing guidance to unravel the complex network of proteomes.
INTRODUCTION
Back in the age of the Roman Empire, fluorescent objects had captured the human imagination. In one of the earliest written accounts of fluorescence, Pliny the Elder, a renowned Roman philosopher, described the fluorescence in jellyfish (Pulmo marinus). This description was later translated by John Bostock and Henry T. Riley in 1855:
“If wood is rubbed with the Pulmo marinus, it will have all the appearance of being on fire; so much so, indeed, that a walking-stick, thus treated, will light the way like a torch" (Pliny, 1855).
With centuries of efforts, the molecular mechanism of fluorescence in marine organisms, such as deep-sea anemones and jellyfishes, is no longer just a subject of fascination but something researchers have come to understand. There have been prominent evolutions in science which diversify the characterisation of fluorescent proteins (FPs), enabling GFP to be an indispensable tool for contemporary research. In this review, we begin by outlining the discovery and physical properties of GFP. The engineering of GFP variants via spectrochemical modulation and folding optimisation is then discussed as optimised FPs can advance the understanding of protein localisations and protein-protein interactions (PPIs). We further summarise additional applications of split-GFP in probing protease activity and protein quantification.
DISCOVERY OF GFP
GFP was first discovered by Osamu Shimomura, 2008 Nobel laureate in Chemistry. Shimomura’s task was to identify the bioluminescent system in Aequorea victoria (O Shimomura, F H Johnson, Y Saiga, 1962) . Extracted and purified from A. victoria, the first protein responsible for the fluorescence was named aequorin. Interestingly, the purified aequorin emitted bluish light, instead of greenish luminescence observed in the light-emitting tissues of A. victoria. Shimomura then postulated the presence of another "green protein" in A. victoria. Later, his team successfully extracted and purified the "green protein" then obtained the respective emission spectra of aequorin, the light-emitting tissue of A. victoria, and this novel "green protein" (Johnson et al., 1962) . Eventually, they confirmed that the fluorescence was emitted by this "green protein" due to the absorption of bluish light from aequorin.
At that time, the mechanism mediating the energy transfer from aequorin to GFP was unclear. This was later elucidated in 1974, suggesting Förster resonance energy transfer (FRET) mediated the energy transfer from aequorin to GFP. Emitted from aequorin, the blue light was readily absorbed by GFP, producing its characteristic fluorescence (Morise et al., 1974) . GFP did not attract much attention in the first few decades after its discovery. It was not until 1994 when Douglas Prasher and his team utilised GFP as a fluorescent tag to report gene expression (Chalfie et al., 1994) that led to the evolution of GFP to be an indispensable research tool nowadays.
PROPERTIES OF GFP: STRUCTURE, MATURATION, AND PHOTOCHEMISTRY

Figure 1. Side view (left) and axial view (right) of GFP's crystal structure. Chromophore is shown in a ball-stick model. Note the unique β-barrel structure of GFP. This enclosed structure is essential for chromophore maturation and GFP fluorescence. This explains why other proteins containing the same Ser-Tyr-Gly motif do not fluoresce (PDB code: 1EMA). Figure was generated using UCSF Chimera.
Structure of GFP
Although the crystal (Morise et al., 1974) and X-ray diffraction pattern (Perozzo et al., 1988) of GFP have been available since 1974 and 1988 respectively, its structure remained a mystery until 1996, when Roger Tsien, 2008 Nobel laureate in Chemistry, and his team unveiled the structure of GFP in Science (Ormö et al., 1996). Wild-type GFP consists of 238 amino acids, with a molecular weight of 26.9kDa. It comprises 11 β-strands twisting and coiling to form a β-barrel, wrapping an α-helix along the axis. The α-helix contains the chromophore of GFP, which is formed from Ser-65, Tyr-66 and Gly-67 (Figure 1). Proper folding of GFP is the key for its fluorescence (Tsien, 1998). The β-barrel encloses the chromophore at the protein core and provides an appropriate microenvironment for fluorescence, protecting the chromophore from fluorescent quenchers such as water, triplet oxygen and photoisomerisation, etc.
Maturation of GFP
To acquire fluorescence, GFP does not rely on external cofactors; rather, the chromophore of GFP must undergo maturation, which is a post-translational modification (PTM) (Tsien, 1998). This is a series of chemical reactions, beginning with nucleophilic cyclisation, followed by dehydration and finally oxidation by atmospheric oxygen, forming p-hydroxybenzylideneimidazolinone (Figure 2). Gly-67 is highly conserved in all mutant forms of GFP, probably because it is the most suitable nucleophile in the cyclisation reaction, whereas Ser-65 and Tyr-66 can be mutated to modulate the spectrochemical properties of GFP (Fu et al., 2015).
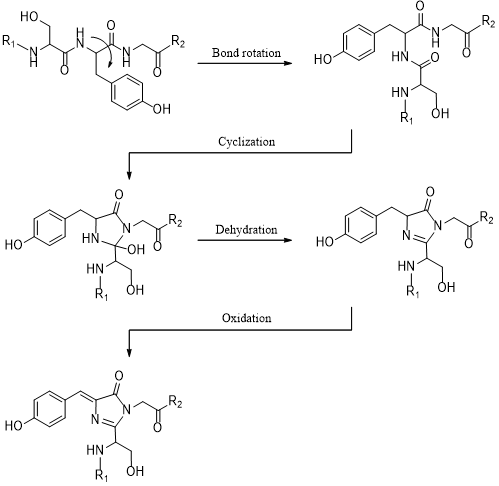
Figure 2. Maturation mechanism of GFP chromophore. Gly-67 is the most critical residue. As the only amino acid with a H atom as its side chain, glycine has remarkable conformational flexibility, permitting its amide nitrogen to attack the carbonyl of Ser-65 at the correct trajectory (Bürgi–Dunitz angle). Thus, Gly-67 is highly conserved among all GFP variants. Figure was generated using ChemDraw.
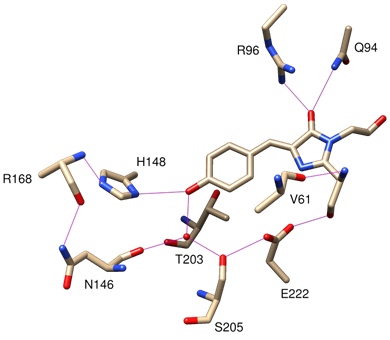
Figure 3. 3D view of the hydrogen bonds between the chromophore and amino acid residues of β-strands. These are crucial for the photochemistry of the chromophore. Note that Thr-203 participates in the solvation of the phenolate chromophore, but not the phenolchromophore. Ionisation of phenol causes the -OH group of Thr-203 to rotate such that the H atom of hydroxyl group can solvate phenolate (PDB code: 1EMA). Figure was generated using UCSF Chimera.
Photochemistry of GFP
In the wild-type chromophore, excitation results in two absorption peaks at 395nm (major) and 475nm (minor) respectively (Tsien, 1998). The fact that GFP has multiple excitation peaks suggests the presence of two distinct photogenic species in the chromophore. Indeed, the chromophore contains a slightly acidic phenol group. Both neutral phenol and anionic phenolate exist in the chromophore. It is estimated the ratio of phenol to phenolate is 6:1 in wild-type GFP. Phenol and phenolate have distinct spectrochemical properties and they are unevenly populated, thereby giving two absorption peaks of different amplitudes upon illumination. The chromophore is tightly held at the core of the β-barrel, stabilised by multiple hydrogen bonds with the side chains of the β strands (Figure 3) (Tsien, 1998).
Excitation of phenol gives a major absorption peak at 395nm, while that of phenolate results in a minor absorption peak at 475nm (Tsien, 1998). Yet relaxation gives a sharp emission peak at 504nm only. This can be explained by excited-state proton transfer (ESPT) (Liu et al., 2018) . Upon excitation, the acidity of phenol increases tremendously. Excited phenol is deprotonated to form phenolate. Thus, the chromophore gives a single emission peak at 504nm, similar to that of excited phenolate.
These unique spectrochemical features of wild-type GFP, however, have several disadvantages for cell biology and protein research. UV excitation (395nm) may cause visual damage to the observer and bleach the tissues/cells to be examined, whereas blue light illumination (475nm) can only excite a small portion of GFP (~15%), which results in very weak fluorescence (Tsien, 1998) . In order to fully utilise GFP in research, extensive protein engineering has been done on it.
PROTEIN ENGINEERING OF GFP
Presented with several limitations, optimisation, and refinement of the wild-type GFP are essential prior to its applications in protein research. Through spectrochemical modulations and folding optimisation, diverse FPs can be engineered to study protein localisations as well as PPIs.
Spectrochemical Modulation
Given the clear understanding of the photochemistry of GFP, it is possible to optimise its spectrochemical properties to meet our needs. For instance, S65T promotes the ionisation of phenol in the chromophore by forcing the carboxyl group Glu-222 to remain in protonated and uncharged state (Jones et al., 2012) . Negatively charged Glu-222 discourages the formation of phenolate anion due to electrostatic repulsion. By transforming the chromophore to phenolate, the absorption peak at 395nm is eliminated. Other mutants can also exhibit various spectrochemical features (Tsien, 1998) . For instance T203Y or other aromatic amino acid substitutions result in π-stacking interaction between the phenolate chromophore and the residue, which stabilises the excited chromophore, thereby causing red shift in both excitation and emission. This created the yellow fluorescent protein (YFP).
Aside from GFP, FPs in other organisms are engineered to produce desired spectrochemical properties. Discovered in Discosoma, red fluorescent protein (RFP) is closely related to GFP based on phylogenetic analysis (Chudakov et al., 2010) . mFruits is a family of FPs derived from RFP. As an example of mFruits, mCherry is characterised by the enormous red shift of its absorption and emission. Rotation of Lys-70 away from the chromophore due to K83L and protonation of Glu-215 are suggested to affect the electron density distribution of the chromophore, leading to a significant red shift, although the exact mechanism is still not clear (Shu et al., 2006) . Additionally, another orange FP called Kusabira-Orange (KO) is derived from DsRed, a monomeric form of RFP (Karasawa et al., 2004) . Rounds of mutagenesis via amino acid substitutions into the AB and AC interfaces of DsRed generates a novel FP — monomeric KO, which has improved folding efficiency, solubility, and brightness (Karasawa et al., 2004).
Apart from RFP, Midori-ishi cyan (MiCy) is a homodimeric cyan fluorescent protein (CFP) originated from Acropara coral (Day and Davidson, 2009; Karasawa et al., 2004). Comparing the amino acid alignment with Aequorea-derived enhanced CFP (ECFP), MiCy carries a Tyr-66 in the second amino acid of the chromophore tripeptide, where ECFP has a Y66W substitution at this position (Karasawa et al., 2004) . Based on spectrochemical analysis, ionised MiCy demonstrates high quantum yield and molar extinction coefficient. Particularly, the fluorescence emitted by MiCy displays a red-shifted profile which has the longest absorption (472nm) and emission wavelength (495nm) among all CFPs. Having a single fluorescence lifetime at a constant 3.4ns, MiCy has its emission spectrum overlaps well with the excitation spectrum of the aforementioned monomeric KO, allowing this donor-acceptor pair suitable for FRET.
With various spectrochemical modulations, the family of GFPs greatly expands. Its family members demonstrate diverse spectrochemical characteristics. Nowadays, it is able to express a rainbow panel of FPs derived from Discosoma RFP and Aequorea GFP (Figure 4) (Shu et al., 2006; Tsien, 2008). As a corollary, molecular biologists can label multiple cellular structures or proteins of interest with FPs at the same time.

Figure 4. Rainbow colours of GFP. (Top) A San Diego beach scene drawn using eight different FPs derived from GFPs and RFPs, created by the laboratory of Roger Tsien. (“San Diego beach scene drawn with an eight colour palette of bacterial colonies expressing fluorescent proteins derived from GFP and red-fluorescent coral protein” by Nathan Shaner is licensed under CC BY-SA 3.0). (Bottom) From left to right: Enhanced blue fluorescent protein (EBFP), ECFP, EGFP, Citnine, mHoneydew, mBanana, mOrange, tdTomato, mTangerine, mStrawberry, mCherry, mGrape-1, mRaspberry, mGrape-2, mPlum.
Folding Optimisation
The folding efficiency of GFP is sensitive to temperature change (Tsien, 1998) . A. victoria inherits mainly in the northwestern Pacific Ocean. The ocean temperature there (9-12ºC) does not pose any challenges for GFP folding. However, mammalian cells/tissues are typically incubated at 37ºC. High temperature may hinder the folding efficiency of GFP. Improperly folded GFP displayed no fluorescence. Thus, it is of paramount significance to produce less temperature sensitive variants of GFP. One way to achieve this is DNA shuffling which allows screening of GFP variants with more desired properties (Tsien, 1998) . Random screenings discovered that F64L improves GFP folding efficiency. Leucine is less bulky than phenylalanine, thereby inducing higher packing efficiency at the core and facilitating GFP folding (Jones et al., 2012) . F64L, together with the previously described S65T, yielded the enhanced GFP (EGFP).

Figure 5. The network of electrostatic interactions formed by alternating positively (R30 and R122) and negatively charged (E32 and E115) residues across β-strands of GFP. Arg-30 is positively charged and large enough to strong electrostatic attractions between Glu-17 and Glu-32. Ser-30 is too small to form the linkage between Glu-32 and Glu-17 and it is neutral (PDB code: 2B3P). Figure was generated using UCSF Chimera.
A more robust folding variant, known as superfolder-GFP (sf-GFP), was later developed (Pedelacq et al., 2006) . This variant of GFP folds well even when conjugated with poorly folded proteins. sf-GFP consists of EGFP mutations (F64L and S65T), cycle-3 mutations (F99S, M153T, and V163A) and also six novel mutations (S30R, Y39N, N105T, Y145F, I171V, and A206V). Cycle-3 mutations reduce the surface hydrophobicity of GFP, thereby lowering its tendency to aggregate (Tsien, 1998) , whereas the six new mutations further promote the folding of GFP (Pedelacq et al., 2006) . Among them, S30R plays the most critical role in enhancing the folding robustness. Arg-30 is a positively charged residue on β-strand 2 and forms a network of electrostatic interactions across β-strands with other negatively charged residues (Figure 5). This network of interactions holds the β-barrel more tightly, thereby facilitating the folding of GFP even under extremely unfavourable conditions.
Split-GFP
A step further would be the engineering of split-GFP from sf-GFP (Cabantous et al., 2005) . Before the advent of split-GFP, the first experiment of split-protein was done in 1957 by Fred Richards (Romei and Boxer, 2019) . His experiment showed that when ribonuclease A was cleaved by subtilisin, two resulting fragments known as S-peptide and S-protein remain tightly bound to one another (Kd = 30pM). Even when ribonuclease S was cleaved, the enzyme was still active. Nonetheless, removal of any of these fragments would abolish its activity. Scientists later discovered some other enzymes also demonstrated these properties. With these characteristics, they designed protein-fragment complementation assay (PCA) to investigate protein localisations and interactions.
The emergence of split-proteins provides a new tool but measuring PPIs remains a challenging task. Most of these split-proteins are enzymes, and thus the only way to verify these interactions is to detect the enzymatic activity by adding a suitable substrate. However, the substrate may not be specific for the split-protein. It can be a substrate for other enzymes as well, giving false positive results. This problem was not well solved until the advent of GFP (Romei and Boxer, 2019) . With its extreme tolerance to circular permutation, any of the β-strands in GFP can become the new N-/C-terminus. Besides, protein insertion to GFP does not affect its functions. Most importantly, protein interactions and localisations can be visualised by the fluorescence of GFP with no substrate or cofactor and the result can be monitored by fluorescent microscopy.

Figure 6. Crystal structure of split-GFP. (Left) Tripartite split-GFP. GFP10 and 11 [forest green and yellow] are detached from GFP1-9. (Right) Bipartite split-GFP. GFP11 [magenta] are detached from GFP1-10 (PDB code: 2B3P). These segments of GFP can be co-expressed with other proteins to detect PPIs. In PCA, when the “bait” and “prey” proteins interact, these segments auto-assemble and GFP fluoresces. Figure was generated using UCSF Chimera.
The advent of GFP opened a new pathway for PCA. GFP can be split into either two (bipartite) or three (tripartite) segments (Figure 6). Bipartite system splits GFP into two parts. The larger segment is GFP1-10, while the smaller one is GFP11 (Dáder et al., 2019) . Tripartite system splits GFP into three parts. The larger segment is GFP1-9, while the smaller are GFP10 and GFP11 (Cabantous et al., 2013) . Only when these segments are in close proximity will they assemble and emit photons. Hence, split-GFP is a promising tool in studying protein localisations and interactions.
Engineering GFPs: An Indispensable Tool in Protein Research
Since the 1990s, GFP has undergone extensive protein engineering to diversify its functions. Years of experiences have proven GFP to be a robust tool in studying proteins. For instance, the localisation of GFP-tagged protein can be tracked in real-time. Split-GFP technology also allows efficient monitoring of different PPIs. In addition, GFP can be applied as an in vivo protein biosensor to report protein activity. All these contribute to a more comprehensive understanding of proteomes.
GFP IN MAPPING PROTEIN LOCATIONS
Comprehensive understanding of a protein’s subcellular locations provides information of protein functions and its possible interacting partner(s). Possible PPIs can be initially observed by co-localisation study. More in-depth assays can then select the proteins of interest to test for interactions. Tagging proteins with GFP has become an important and convenient strategy for studying protein localisations.
Mapping of Global Protein Localisations with GFP Tags
A study in 2003 performed a large-scale analysis of protein localisations in the model yeast S. cerevisiae (Huh et al., 2003). Through oligonucleotide-directed homologous recombination, 4156 proteins, which constitute 75% of the yeast proteome, were tagged with GFP for localisation detected by fluorescence microscopy. By combining results from the mass localisation of yeast proteins and a database of pre-existing PPIs known as GRID (Rédei, 2008), significant correlations between protein localisations and PPIs were found. The study visually linked distinct subcellular locations with PPIs, suggesting proteins interact preferentially based on their partner’s subcellular origin (Figure 7). This study, while uncovering novel locations of yeast proteins with GFP, allowed implications of PPIs and functions from their locations. Hence, future proteomic researchers can predict, with certain confidence, the functions and interacting partners of novel proteins based on their subcellular locations.

Figure 7. A diagram of linkages between subcellular components with statistically significant PPIs. Proteins from respective linked subcellular components have a higher propensity to interact with each other. Figure was generated using ChemDraw.
In 2011, an investigation on Caenorhabditis elegans (C. elegans) was conducted in a similar fashion (Meissner et al., 2011). With their proportionally large body wall muscle cells along with the abundance of protein homologs to human muscle proteins, performing research on C. elegans has given considerable advantages for the study of skeletal muscle diseases. Gateway recombination cloning system (Walhout et al., 2000) was applied in tagging proteins in C. elegans body wall muscle cells with GFP, most of which are orthologs of human proteins. The localisations of 227 proteins were identified and subsequently assigned to categories 1 to 15 based on their localisation pattern. At least 80 proteins appeared to be novel components in known muscle specific structures, a few of which (D2092.4, F42C5.9, K06A4, and R11G1) exhibit unique and unusual localisation features. With novel locations for proteins of unknown functions, possible functions and interacting partners could be narrowed down. The global localisation of orthologs could hence serve as an invaluable resource in the investigation on human sarcomere assembly and function.
Monitoring the Localisation of Virulence Factors by Split-GFP
Apart from the applications of GFP in the mass localisation of yeast proteins and C. elegans, split-GFP, as an advanced GFP variant, demonstrated the ability to locate viral virulence factors with high specificity as illustrated by viral 3A protein and bacterial internalin C (InlC) protein in the following.
Localisation of 3A Protein of Coxsackievirus B3 in Infected Cells
During infection, the inner cellular structures of host cells are reformed to create sites for viral RNA replication (Boon and Ahlquist, 2010) . The tubular structures formed from remodelled Golgi membranes are known as replicating organelles (ROs), which are essential for viral replication (Belov et al., 2011).
A sf-GFP-based bipartite system was used to tag the 3A protein of coxsackievirus B3 (CVB3) (van der Schaar et al., 2016) , which is a small protein known to insert into RO membranes during CVB3 infection (Towner et al., 1996) . The sf-GFP is split into a small segment (GFP11) and a large segment (GFP1-10). Shown to accept small epitope tags without disturbing its function (Teterina et al., 2011) , viral 3A was tagged with GFP11 while infected host cells expressed GFP1-10. Both segments are non-fluorescent on their own but fluoresce when combined within the host cell in order to locate 3A proteins.

Figure 8. Confocal microscopy of Buffalo Green Monkey cells (GFP1-10) transduced with mCherry-GM130 infected with CVB3 (GFP11). Green fluorescence from GFP indicates the emergence of 3A proteins, while fading red fluorescence from mCherry indicates dissociation of Golgi. (Reproduced with permission of van der Schaar et al., 2016)
Real-time live-cell imaging was applied to localise 3A proteins (van der Schaar et al., 2016). While tagging 3A with GFP11, GM130, a protein marker for the host cell Golgi apparatus, was transduced with traceable red marker mCherry (Figure 8). As expected, fluorescence from GFP11-tagged 3A slowly emerged over time, while red fluorescence of mCherry-GM130 faded during the infection process, indicating the emergence of 3A dissociates the Golgi apparatus. As seen from live microscopy, split-GFP could reveal the dynamic locations of 3A in real time in relation to proteins affected by the infection.
As demonstrated by the study, split-GFP provided accurate localisation of tagged 3A without significant alterations in protein functions. The development of viral-induced structures also remained unperturbed. A smaller tag (GFP11) inferred less bulk on 3A which prevented obstructions in protein interactions. Moreover, split-GFP allowed the localisation of 3A in real time, which revealed dynamic information previously unattainable with static immunoassays.
Localisation of Internalin C of Listeria monocytogenes in Host Cells
In addition to its applications on molecular virology, split-GFP serves as a modular platform to visualise myriads of virulence proteins secreted by pathogenic bacteria (Batan et al., 2018; Mcquate et al., 2017; Young et al., 2017). Recently, an engineered multicolour split-GFP has been designed to monitor the accumulation and distribution of virulence proteins secreted by Listeria monocytogenes within the host cell (Batan et al., 2018).
As a foodborne pathogen causing listeriosis, Listeria secretes diverse virulence proteins which create heterogenous phenotypes between individual cells upon infection (Batan et al., 2018; Helaine and Holden, 2013). Understanding the subcellular localisation and spatial-temporal expression of these virulence proteins can provide insights into the progression of Listeria infection.
Although secreted Listeria virulence proteins have been routinely studied by static immunofluorescence, no biochemical assay was demonstrated to evaluate the dynamics of virulence proteins with respect to the infection of live Listeria or other Gram-positive bacteria (Batan et al., 2018; Kühbacher et al., 2013; Moseley et al., 2007). This hampers researchers to comprehensively investigate the dynamic localisation and functions of the virulence proteome. Hence, proteins of interest can be labelled by different fluorescent tags and visualised by split-GFP. D Batan, E Braselmann, M Minson, D Nguyen, P Cossart, A Palmer (2018) have applied split-GFP to visualise the localisation of internalin C (InlC), a virulence Listeria protein.

Figure 9. Crystal structure of fusion InlC-GFP. The C-terminus of InlC [cyan] is linked to the N-terminus of GFP11 [orange] with a flexible linker. InlC-GFP11 complements with GFP1-10 [blue] to produce fluorescent InlC-GFP. The structure is combined from two separate models (PDB codes: 1XEU and 1GFL). Figure was generated using UCSF Chimera.
InlC was found to interfere with host innate immune response via interaction with IκB kinase alpha (IKKα) (Gouin et al., 2010) and facilitate cell-to-cell dissemination by minimising cortical tension (Rajabian et al., 2009) . To track the secretion dynamics of InlC, analogous to the process in tagging 3A protein of CVB3, the split-GFP was initially engineered from sf-GFP (Feng et al., 2017) in which the non-fluorescent GFP11 fragment was fused to the C-terminus of InlC with a flexible linker (Figure 9) (Batan et al., 2018; Polle et al., 2014). The remaining segment GFP1-10 is non-fluorescent and expressed in mammalian host cells. During Listeria infection, GFP11 of the secreted InlC-GFP11 complex complements with GFP1-10, constituting the complemented fluorescent InlC-GFP for visualisation of InlC dynamics in the infected cells (Figure 10) (Batan et al., 2018).

Figure 10. General strategy for tagging Listeria virulence proteins with bipartite split-GFP. Host cell was firstly transfected with a DNA encoding nonfluorescent GFP1-10 fragment. Listeria cells were electroporated with pAT18 plasmids expressing nonfluorescent fusion InlC-GFP11 protein. Host cells were then infected with these Listeria cells. Upon secretion of InlC-GFP11, two GFP fragments complement to produce a fluorescent GFP.
The next step to understand protein localisations from split-GFP would be establishing multicoloured fluorescent imaging strategies. Since the Listeria expression plasmid is modular, split-fluorescent protein tags can be exchanged easily (Batan et al., 2018) . Notably, GFP11 tag can be exchanged with split mNeon-Green11 and split superfolder Cherry11. All these variants resemble the complementation of GFP1-10 and InlC-GFP11 described above, allowing the generation of multicoloured fluorescence when mNeon-Green1-10 or superfolder Cherry1-10 is used, respectively (Batan et al., 2018) . This flexibility enables us to evaluate the secretory dynamics of virulence proteins at single cell level, which are fundamental to dissect the proteome function and sophisticated infection process in order to formulate the most appropriate treatment regimen.
Nonetheless, one major concern when implementing split-GFP on protein localisation studies is whether the tagged protein still retains its original properties and functions. Despite its small size, GFP11 may still affect the functions of the tagged proteins. Thus, it is necessary to evaluate if the tagged protein retains its functions alongside with the localisation study.
GFP IN UNDERSTANDING PROTEIN-PROTEIN INTERACTIONS
Besides protein localisations, PPIs are also important in protein research. Many crucial cellular processes, namely signal transductions, immune responses, etc. are mediated by PPIs. Split-GFP has an enormous potential to be a tool to study such interactions. The aforementioned bipartite split-GFP system, nonetheless, bears weaknesses. Bipartite split-GFP constitutes a small and large segment. When separately linked to proteins, the large segment may disturb the functions of the tagged protein. Furthermore, it may undergo self-assembly, leading to false-positive results.
Direct Protein Association
To further improve on the bipartite system, tripartite split-GFP is developed for accurate measurement of PPIs (Cabantous et al., 2013). A tripartite split-GFP is segregated into three components. Two small segments, GFP10 and GFP11, are fused to two proteins-of-interest. When these fusion proteins interact, GFP10 and GFP11 are tethered due to their close proximity. For the emission of GFP fluorescence, GFP1-9 must then fuse with GFP10 and GFP11 to form a complete GFP. If both fusion proteins do not interact, GFP10 and GFP11 do not assemble. No fluorescence will be observed even with the addition of GFP1-9 as the entropy penalty is too high for the complementation of three separate components. A flowchart briefly illustrates the above process (Figure 11).

Figure 11. Fluorescent-emitting mechanismof tripartite split-GFP system. Protein A and Protein B are fused to GFP10 or GFP11, respectively. Protein A interacts with Protein B, bringing two small GFP fragments (GFP10 and GFP11) into close proximity. Under the co-expression of GFP1-9, all these GFP fragments reconstitute to form a functional GFP. Figure was generated using ChemDraw.
To validate the effectiveness of tripartite GFP in detecting direct protein association, coiled- coil domains are used to test fluorescent levels depending on whether PPI is present or not (Cabantous et al., 2013; Tripet et al., 1997). Two sets of lysine-rich (K1) and glutamate-rich (E1) coiled-coil domains were employed. The oppositely charged K1/E1 pair undergoes hetero-dimerisation, while the negatively charged E1/E1 pair cannot dimerise owing to electrostatic repulsion. Both proteins in each set were tagged with either GFP10 or GFP11 and transformed into GFP1-9 expressing E. coli. E. coli co-expressing K1/E1 turned brightly fluorescent due to K1/E1 interactions. In contrast, cells with E1/E1 produced baseline fluorescence comparable to negative control levels, suggesting minimal GFP10-GFP11 self-assembly. The data suggests high effectiveness of the tripartite system in visualising PPIs with few false positive results.
Apart from the aforementioned measurement of direct PPIs conducted in E. coli, the tripartite system has also been involved in detection of membrane PPIs in Arabidopsis (Liu et al., 2018) . The application of tripartite split-GFP on detecting membrane PPIs involved in phosphate homeostasis provides results with high fidelity. The tripartite system was also successfully transformed within various cellular components in planta. Thus, the tripartite system demonstrates considerable diversity and extends its applications to proteins within plant cells.
Induced Protein Interactions
In addition to direct protein association, this system remains effective in monitoring induced PPIs. Rapamycin, a well-studied immunosuppressant, induces FKBP12-FRB binding (Banaszynski et al., 2005). The tripartite system was applied to measure fluorescence from FKBP12-FRB binding in rapamycin positive and negative groups as illustrated (Cabantous et al., 2013) (Figure 12). Similar to previously described methods, FRB-GFP10 and FKBP12-GFP11 were co-expressed in E. coli. GFP1-9 was then added to the crude extracts of E. coli, along with rapamycin for the positive group and none for the negative group. A two-fold increase in fluorescence was detectable after 10 minutes, followed by a continuous increase in the presence of rapamycin, while the absence of rapamycin resulted in nearly blank levels. This illustrates the successful detection of drug-induced PPI, which opens up possibilities of tripartite GFP in evaluating small molecule-mediated PPIs. It should be noted, although GFP can monitor small molecule-mediated PPIs, it cannot be directly tagged to small molecules for localisation and interaction studies. In fact, due to the nature of GFP as a protein instead of a small fluorescent molecule, it is only suitable for tagging DNA-encoded biomolecules, i.e., proteins.

Figure 12. Schematic diagram showing rapamycin-induced FRB-FKBP12 interactions, which can be detected by tripartite split-GFP complementation. The diagram shows the mechanism of rapamycin-mediated FRB-GFP10 and FKBP12-GFP11 interaction, followed by GFP1-9 complementation and emission of GFP fluorescence. Figure was generated using ChemDraw.
Indirect Protein Associations
Apart from direct or drug-mediated PPIs, the assembly of protein complexes has been a challenge for GFP-tag systems as complexes often involve more than two interacting proteins. To examine whether the tripartite system can detect the assembly of complexes or not, the Tus BCD complex (YheNML) from E. coli was used (Cabantous et al., 2013). YheM protein acts as a bridge between YheN and YheL to form the YheNML heterotrimer. Indirect interaction between YheN and YheL is only possible in the presence of YheM. YheN-GFP10 and YheL-GFP11 are co-expressed in E. coli, followed by complementation of GFP1-9 (Figure 13). High levels of fluorescence were observed in colonies with complete YheNML expression. Weak fluorescence was detected in YheM and YheL only cells. This shows the tripartite system has the ability to detect even indirect PPIs in multi-subunit complexes (Pedelacq and Cabantous, 2019).
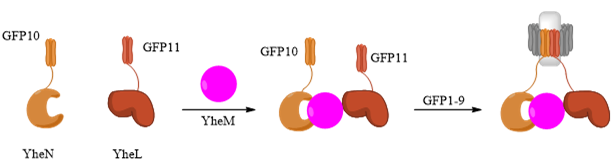
Figure 13. Schematic diagram showing indirect protein association of YheNML, which can be detected by tripartite split-GFP complementation. The diagram shows the mechanism of YheM-mediated YheN-GFP10 and YheL-GFP11 interaction, followed by GFP1-9 complementation and emission of GFP fluorescence. Figure was generated using ChemDraw.
OTHER APPLICATIONS OF GFP IN PROTEIN RESEARCH
Having seen how split-GFP helps in understanding PPIs, the flexibility of engineering split-GFP advances extra applications of GFP in cell biology and protein research.
GFP as Protease Reporter
A novel application of GFP is to probe the activity of proteases in vivo. To achieve this, the concept of “FlipGFP” is introduced (Zhang et al., 2019). As the name “flip” suggests, the orientation of GFP10 and GFP11 is inverted. To generate this conformation, two peptide motifs, E5 and K5, are incorporated into them. E5 is inserted between GFP10 and GFP11, whereas K5 is linked to the C-terminus of GFP11. Protease cleavage sequence is inserted between GFP11 and K5 (Figure 14A). Once this construct is expressed, E5 and K5 dimerise. Dimerisation inverts the orientation of GFP11 such that it becomes parallel, instead of antiparallel, to GFP10. This parallel conformation prevents its complementation with GFP1-9. Upon protease cleavage, GFP11 can freely rotate. GFP10 and 11 can then assemble to GFP1-9 in an antiparallel manner (Figure 14B). A 77-fold increase in fluorescence was recorded (Zhang et al., 2019).

Figure 14. “FlipGFP” as a protease activity reporter. (A) Sensor construct containing GFP10, GFP11 and E5/K5 coiled-coil domains. (B) E5/K5 inverts the orientation of GFP11. This conformation blocks their complementation with GFP1-9. Protease cleavage frees up GFP11, which align anti-parallelly with GFP10, allowing the reconstitution of the functional GFP. Figure was generated using ChemDraw.
In 2019, a group of scientists successfully utilised this strategy to report the activity of endogenous caspase 3, a critical executioner caspase in apoptosis (Zhang et al., 2019). This allowed real-time imaging of apoptotic cells and facilitated our understanding of caspases in vivo. The researchers also examined the feasibility of using FlipRFP as a protease activity reporter. FlipRFP was found to demonstrate similar activity as FlipGFP, and therefore, in theory, the idea of FlipGFP can be applied as a biosensor to detect novel proteases, especially when a tag may interfere with their functions.
Protein Quantification with GFP
Bipartite split-GFP system can also be applied to quantify proteins. First tagged with GFP11, the engineered protein-of-interest is expressed, followed by cell lysis. The cell lysate obtained is then treated with recombinant GFP1-10 (as a reagent). The complementation of GFP1-10 and GFP11 tagged protein-of-interest produces green fluorescence. The intensity of fluorescence can be used to quantify the protein-of-interest (Pedelacq and Cabantous, 2019).
As an illustration, the aggregation of microtubule-associated Tau proteins as neurofibrillary tangles, which are pathologically associated with Alzheimer’s and Parkinson’s disease, can be measured quantitatively with this bipartite split-GFP complementation assay (Chun et al., 2007) . Tau protein was fused to GFP11 and this fusion protein is prone to aggregation under PTMs such as caspase 3-mediated cleavage and abnormal pseudophosphorylations at Ser-396 and Ser-404. Aggregated Tau loses the ability for GFP reconstitution because its GFP11 tag has been sequestered intramolecularly. To monitor the aggregation of Tau in situ, the intensity of GFP fluorescence decreases with the extent of Tau aggregation, reflecting this system is feasible in determining the Tau aggregation process in living mammalian cells.
Furthermore, this split-GFP assay allows researchers to quantify the solubility of wild-type and artificially re-designed α-synuclein (Kothawala et al., 2012) . α-synuclein, which constitutes the major component of Lewis bodies in neurons, is a misfolding-prone protein with high propensity to aggregation in Parkinson’s disease. The traditional method to visualise protein aggregation in living cells was performed by overexpressing α-synuclein fused to a GFP reporter. However, any aggregation events that occur after the formation of GFP chromophore do not affect the emission of fluorescence. In other words, the detection of GFP fluorescence cannot reflect the aggregation state of α-synuclein. In the refined split-GFP assay, α-synuclein was fused to the “sensor” GFP11 fragment while the “detector” GFP1-10 fragment was co-expressed in HeLa cell cultures. The intensity of GFP fluorescence is directly proportional to the solubility of α-synuclein because fluorescence is emitted only when the sensor fragment is escaped from aggregation and complements with the detector fragment. All of these studies present the important utility of split-GFP assays in quantifying proteins, thus broadening the understanding of neurodegenerative diseases.
CONCLUSION AND FUTURE PERSPECTIVES
Through exploring the discovery, structure, and maturation mechanisms of GFP, the scientific community has optimised GFP and developed variants to fit specific needs in conducting biomedical research. Early variants of GFP have been crucial for localising proteins as demonstrated in the global analysis of protein localisation in yeast (Huh et al., 2003) and C. elegans (Meissner et al., 2011) , thereby predicting the functions of novel proteins. The use of split-GFP has further propagated the visualisation of proteins in real-time. With the emergence of multicolour split-GFP, virulence proteins that previously relied on static immunoassays can now be tracked dynamically. With further improvement of bipartite split-GFP, tripartite split-GFP was designed to tag up to two proteins with GFP10 and GFP11 subunits, creating an ideal method for visualising PPIs. This facilitates the elucidation of protein pathways and generates new methods for "hit-to-lead" by monitoring drug-induced PPIs. Visualisation aside, GFP’s applications extend towards the measurement of protease activity and protein quantification, showing a high degree of versatility when engineered with novel modifications.
Given the ability of GFP in undergoing modifications, the prospects of GFP may involve refinements of existing variants, discovery of novel variants or even fusion of different variants. Refinements of existing variants have been previously observed with sf-GFP to generate stronger and more stable fluorescence. Also, extending the emission spectra of FPs, such as far-red mNeptune with an excitation peak at 600nm and an emission peak at 650nm, can promote the study of PPIs in living cells and mice with excellent signal-to-noise ratios (Han et al., 2014; Pedelacq and Cabantous, 2019). Additionally, the maturation rate of GFP chromophore after complementation can be improved in several ways: First, better understanding of the folding of monomeric GFP1-10 fragment improves its complementation process with GFP11 (Köker et al., 2018; Pedelacq and Cabantous, 2019). Second, the application of GFP binders such as camelid-derived single heavy-chain antibodies (i.e. VHH or nanobodies) can reversibly modulate the GFP fluorescence (Kirchhofer et al., 2010). Nanobodies induce rearrangements of the GFP environment (Kirchhofer et al., 2010). Technically, the binding of nanobodies induces a conformational shift of the loop region from Glu-142GFP to His-148GFP, allowing Arg-168GFP in close proximity with His-148GFP. The side chain of Arg-168GFP is then stabilised by direct interactions with Tyr-37 and Glu-101 on the nanobody so that the proton acceptor His-148GFP is held close to the GFP chromophore. Co-localisation studies revealed such anti-GFP nanobodies bind exclusively to tripartite reconstituted GFP, rather than the individual GFP1-9 fragment (Koraïchi et al., 2018) . The mechanism of how nanobodies enhance GFP fluorescence remains unclear, possibly because the nanobody stabilises the interaction between amino acid residues on both GFP and nanobody, contributing to the stable formation of chromophore (Pedelacq and Cabantous, 2019). Modern refinements, such as acid-tolerant GFP variations, will pinpoint and overcome current weaknesses of GFP (Shinoda et al., 2018). Generation of novel GFP variants will become more practical and efficient with the recent emergence of gene-editing tools. The concept of integrating GFP variants, such as the integration of tripartite split-GFP with light-regulated GFP has also been conducted (Do and Boxer, 2011) . These mentioned modifications can hopefully be applied in future proteomic studies to create tailor-made GFPs, which serve to unravel the mystery of complex proteomes.
CONFLICTS OF INTEREST/DISCLOSURE
The authors declare no conflicts of interest.
ACKNOWLEDGEMENTS
We would like to express our gratitude to Dr. Masayo Kotaka for her guidance throughout the process of idea refinement and manuscript writing. We would also like to thank all anonymous reviewers for their constructive comments on refining this article.
REFERENCES
Banaszynski, L.A., Liu, C.W. and Wandless, T.J. (2005) “Characterization of the FKBP·Rapamycin·FRB Ternary Complex,” Journal of the American Chemical Society, 127(13), 4715–4721, available: doi: 10.1021/ja043277y.
Batan, D., Braselmann, E., Minson, M., Nguyen, D., Cossart, P. and Palmer, A. (2018). A Multicolor Split-Fluorescent Protein Approach to Visualize Listeria Protein Secretion in Infection. Biophysical Journal, 115(2), 251-262.
Belov, G. A., Nair, V., Hansen, B. T., Hoyt, F. H., Fischer, E. R. and Ehrenfeld, E. (2011). Complex Dynamic Development of Poliovirus Membranous Replication Complexes. Journal of Virology, 86(1), 302–312.
Boon, J. A. D. and Ahlquist, P. (2010). Organelle-Like Membrane Compartmentalization of Positive-Strand RNA Virus Replication Factories. Annual Review of Microbiology, 64(1), 241–256.
Cabantous, S., Nguyen, H. B., Pedelacq, J. D., Koraïchi, F., Chaudhary, A., Ganguly, K., Lockard, M. A., Favre, G., Terwilliger, T. C. and Waldo, G. S. (2013). A New Protein-Protein Interaction Sensor Based on Tripartite Split-GFP Association. Scientific Reports, 3(1), available: doi: 10.1038/srep02854.
Cabantous, S., Terwilliger, T. C. and Waldo, G. S. (2005). Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nature biotechnology, 23(1), 102-107.
Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. and Prasher, D. (1994). Green fluorescent protein as a marker for gene expression. Science, 263(5148), 802-805.
Chudakov, D. M., Matz, M. V., Lukyanov, S. and Lukyanov, K. A. (2010). Fluorescent proteins and their applications in imaging living cells and tissues. Physiological Reviews, 90(3), 1103-1163.
Chun, W., Waldo, G. S. and Johnson, G. V. (2007). Split GFP complementation assay: a novel approach to quantitatively measure aggregation of tau in situ: effects of GSK3β activation and caspase 3 cleavage. Journal of Neurochemistry, 103(6), 2529-2539.
Dáder, B., Burckbuchler, M., Macia, J. L., Alcon, C., Curie, C., Gargani, D., Zhou, J. S., Ng, J. C. K., Brault, V. and Drucker, M. (2019). Split green fluorescent protein as a tool to study infection with a plant pathogen, Cauliflower mosaic virus. PloS one, 14(3), e0213087.
Day, R. N. and Davidson, M. W. (2009). The fluorescent protein palette: tools for cellular imaging. Chemical Society Reviews, 38(10), 2887-2921.
Do, K. and Boxer, S. G. (2011). Thermodynamics, Kinetics, and Photochemistry of β-Strand Association and Dissociation in a Split-GFP System. Journal of the American Chemical Society, 133(45), 18078–18081, available: doi: 10.1021/ja207985w.
Feng, S., Sekine, S., Pessino, V., Li, H., Leonetti, M. and Huang, B. (2017). Improved split fluorescent proteins for endogenous protein labeling. Nat Commun, 8(1), 370.
Fu, J. L., Kanno, T., Liang, S., Matzke, A. J. and Matzke, M. (2015). GFP loss-of-Function mutations in Arabidopsis thaliana. G3: Genes|Genomes|Genetics, 5(9), 1849-1855.
Gouin, E., Adib-Conquy, M., Balestrino, D., Nahori, M., Villiers, V., Colland, F., Dramsi, S., Dussurget, O. and Cossart, P. (2010). Listeria monocytogenes InlC protein interferes with innate immune responses by targeting the IκB kinase subunit IKKα. Proceedings of the National Academy of Sciences of the United States of America, 107(40), 17333-17338.
Han, Y., Wang, S., Zhang, Z., Ma, X., Li, W., Zhang, X., Deng, J., Wei, H., Li, Z., Zhang, X. and Cui, Z. (2014). In vivo imaging of protein–protein and RNA–protein interactions using novel far-red fluorescence complementation systems. Nucleic acids research, 42(13), e103-e103.
Helaine, S. and Holden, D. (2013). Heterogeneity of intracellular replication of bacterial pathogens. Current Opinion in Microbiology, 16(2), 184-191.
Huh, W. K., Falvo, J. V., Gerke, L. C., Carroll, A. S., Howson, R. W., Weissman, J. S. and O’Shea, E. K. (2003) Global analysis of protein localization in budding yeast. Nature, 425, 686–691.
Johnson, F. H., Shimomura, O., Saiga, Y., Gershman, L. C., Reynolds, G. T. and Waters, J. R. (1962). Quantum efficiency of Cypridina luminescence, with a note on that of Aequorea. Journal of Cellular and Comparative Physiology, 60(1), 85-103.
Jones, D., Arpino, J. and Rizkallah, P. (2012). Crystal structure of enhanced green fluorescent protein to 1.35a resolution reveals alternative conformations for Glu222. PLoS ONE, 7(10), 1849-1855.
Karasawa, S., Araki, T., Nagai, T., Mizuno, H. and Miyawaki, A. (2004). Cyan-emitting and orange-emitting fluorescent proteins as a donor/acceptor pair for fluorescence resonance energy transfer. Biochemical Journal, 381(1), 307-312.
Kirchhofer, A., Helma, J., Schmidthals, K., Frauer, C., Cui, S., Karcher, A., Pellis, M., Muyldermans, S., Casas-delucchi, C. S., Cardoso, M. C., Leonhardt, H., Hopfner, K. and Rothbauer, U. (2010). Modulation of protein properties in living cells using nanobodies. Nature Structural & Molecular Biology, 17(1), 133.
Köker, T., Fernandez, A. and Pinaud, F. (2018). Characterization of split fluorescent protein variants and quantitative analyses of their self-assembly process. Scientific Reports, 8(1), 1-15.
Koraïchi, F., Gence, R., Bouchenot, C., Grosjean, S., Lajoie-Mazenc, I., Favre, G. and Cabantous, S. (2018). High-content tripartite split-GFP cell-based assays to screen for modulators of small GTPase activation. Journal of Cell Science, 131(1).
Kothawala, A., Kilpatrick, K., Novoa, J. A. and Segatori, L. (2012). Quantitative analysis of α-synuclein solubility in living cells using split GFP complementation. PLoS One, 7(8), e43505.
Kühbacher, A., Gouin, E., Mercer, J., Emmenlauer, M., Dehio, C., Cossart, P. and Pizarro-Cerdá, J. (2013). Imaging InlC secretion to investigate cellular infection by the bacterial pathogen Listeria monocytogenes. Journal of Visualized Experiments : JoVE, (79), E51043.
Liu, M., Yu, X., Li, M., Liao, N., Bi, A., Jiang, Y., Liu, S., Gong, Z. and Zeng, W. (2018). Fluorescent probes for the detection of magnesium ions (Mg2+): From design to application. RSC Advances, 8(23), 12573-12587.
Liu, T. Y., Chou, W. C., Chen, W. Y., Chu, C. Y., Dai, C. Y. and Wu, P. Y. (2018). Detection of membrane protein–protein interaction in planta based on dual‐intein‐coupled tripartite split‐GFP association. The Plant Journal, 94(3), 426–438, available: doi: 10.1111/tpj.13874
McQuate, S. E., Young, A. M., Silva‐Herzog, E., Bunker, E., Hernandez, M., de Chaumont, F., Liu, X., Detweiler, C. S. and Palmer, A. E. (2017). Long‐term live‐cell imaging reveals new roles for Salmonella effector proteins SseG and SteA, Cellular Microbiology, 19(1), e12641.
Meissner, B., Rogalski, T., Viveiros, R., Warner, A., Plastino, L., Lorch, A., Granger, L., Segalat, L. and Moerman, D. G. (2011). Determining the Sub-Cellular Localization of Proteins within Caenorhabditis elegans Body Wall Muscle. PLoS ONE, 6(5), available: doi: 10.1371/journal.pone.0019937
Morise, H., Shimomura, O., Johnson, F. H. and Winant, J. (1974). Intermolecular energy transfer in the bioluminescent system of Aequorea. Biochemistry, 13(12), 2656-2662.
Moseley, F., Bicknell, K., Marber, M. and Brooks, G. (2007). The use of proteomics to identify novel therapeutic targets for the treatment of disease. Journal of Pharmacy and Pharmacology, 59(5), 609-628.
Ormö, M., Cubitt, A. B., Kallio, K., Gross, L. A., Tsien, R.Y. and Remington S. J. (1996). Crystal structure of the Aequorea victoria green fluorescent protein. Science, 273(5280), 1392-5.
Pedelacq, J. and Cabantous, S. (2019). Development and applications of Superfolder and split fluorescent protein detection systems in biology. International Journal of Molecular Sciences, 20(14), 3479.
Pedelacq, J., Cabantous, S., Tran, T., Terwilliger, T. C. and Waldo, G. S. (2006). Engineering and characterization of a superfolder green fluorescent protein. Nature Biotechnology, 24(9), 1170-1170.
Perozzo, M. A., Ward, K. B., Thompson, R. B. and Ward, W. W. (1988). X-ray diffraction and time-resolved fluorescence analyses of Aequorea green fluorescent protein crystals. Journal of Biological Chemistry, 263(16), 7713-7716.
Pliny. (1855) “Chapter 52 - Other aquatic productions. Adarca or Calamochnos: three remedies. Reeds: eight remedies. The ink of the sæpia.,” in Bostock, J. and Riley, H.T., trans., The Natural History of Pliny, London: H. G. Bohn, 58–59.
Polle, L., Rigano, L., Julian, R., Ireton, K. and Schubert, W. (2014). Structural Details of Human Tuba Recruitment by InlC of Listeria monocytogenes Elucidate Bacterial Cell-Cell Spreading. Structure, 22(2), 304-314.
Rajabian, T., Gavicherla, B., Heisig, M., Müller-Altrock, S., Goebel, W., Gray-Owen, S. D. and Ireton, K. (2009). The bacterial virulence factor InlC perturbs apical cell junctions and promotes cell-to-cell spread of Listeria. Nature cell biology, 11(10), 1212-1218.
Rédei (2008). GRID (general repository for protein interaction database). Encyclopedia of Genetics, Genomics, Proteomics and Informatics, 825–825, available: doi: 10.1007/978-1-4020-6754-9_7153.
Romei, M. G. and Boxer, S. G. (2019). Split green fluorescent proteins: scope, limitations, and outlook. Annual Review of Biophysics, 48(1), 19-44.
Shimomura, O., Johnson, F. H. and Saiga, Y. (1962). Extraction, purification and properties of Aequorin, a bioluminescent protein from the luminous Hydromedusan, Aequorea. Journal of Cellular and Comparative Physiology, 59(3), 223-239.
Shinoda, H., Ma, Y., Nakashima, R., Sakurai, K., Matsuda, T. and Nagai, T. (2018). Acid-Tolerant Monomeric GFP from Olindias formosa. Cell Chemical Biology, 25(3), available: doi: 10.1016/j.chembiol.2017.12.005.
Shu, X., Shaner, N. C., Yarbrough, C. A., Tsien, R. Y. and Remington, S. J. (2006). Novel chromophores and buried charges control color in mFruits†,‡. Biochemistry, 45(32), 9639-9647.
Teterina, N. L., Pinto, Y., Weaver, J. D., Jensen, K. S. and Ehrenfeld, E. (2011). Analysis of Poliovirus Protein 3A Interactions with Viral and Cellular Proteins in Infected Cells. Journal of Virology, 85(9), 4284–4296.
Towner, J. S., Ho, T. V. and Semler, B. L. (1996). Determinants of Membrane Association for Poliovirus Protein 3AB. Journal of Biological Chemistry, 271(43), 26810–26818, available: doi: 10.1074/jbc.271.43.26810.
Tripet, B., Yu, L., Bautista, D. L., Wong, W. Y., Irvin, R. T. and Hodges, R. S. (1997). Engineering a de novo designed coiled-coil heterodimerization domain for the rapid detection, purification and characterization of recombinantly expressed peptides and proteins. Protein Engineering Design and Selection, 10(3), 299–299, available: doi: 10.1093/protein/10.3.299.
Tsien, R. Y. (1998). The green fluorescent protein. Annual Review of Biochemistry, 67, 509-44.
Tsien, R. Y. (2008) Constructing and Exploiting the Fluorescent Protein Paintbox [online], Nobel Lecture, available: https://www.nobelprize.org/prizes/chemistry/2008/tsien/lecture/ [accessed 12 Aug 2020].
van der Schaar, H., Melia, C., van Bruggen, J., Strating, J., van Geenen, M., Koster, A., Bárcena, M. and van Kuppeveld, F. (2016). Illuminating the Sites of Enterovirus Replication in Living Cells by Using a Split-GFP-Tagged Viral Protein. mSphere, 1(4).
Walhout, A. J., Temple, G. F., Brasch, M. A., Hartley, J. L., Lorson, M. A., Heuvel, S. V. and Vidal, M. (2000). [34] GATEWAY recombinational cloning: Application to the cloning of large numbers of open reading frames or ORFeomes. Methods in Enzymology Applications of Chimeric Genes and Hybrid Proteins - Part C: Protein-Protein Interactions and Genomics, available: doi: 10.1016/s0076-6879(00)28419-x.
Young, A., Minson, M., McQuate, S. and Palmer, A. (2017). Optimized Fluorescence Complementation Platform for Visualizing Salmonella Effector Proteins Reveals Distinctly Different Intracellular Niches in Different Cell Types. ACS Infectious Diseases, 3(8), 575-584.
Zhang, Q., Schepis, A., Huang, H., Yang, J., Ma, W., Torra, J., Zhang, S., Yang, L., Wu, H., Nonell, S., Dong, Z., Kornberg, T. B., Coughlin, S. R. and Shu, X. (2019). Designing a green Fluorogenic protease reporter by flipping a beta Strand of GFP for imaging Apoptosis in animals. Journal of the American Chemical Society, 141(11), 4526-4530.