

DANIEL PHILLIPS, WENBIN GUO, RUNXUAN ZHANG
School of Biological Sciences, University of Aberdeen, Aberdeen, United Kingdom
Plant Sciences Division, School of Life Sciences, University of Dundee, Dundee, United Kingdom
Information and Computational Sciences, The James Hutton Institute, Dundee, United Kingdom
ABSTRACT
Post-transcriptional, transcript-level regulation of gene expression through mechanisms like differential alternative splicing is increasingly demonstrated to play a central role in the regulation of many important biological phenomena. However, investigation of such activity is often absent in transcriptomic studies, partly due to a lack of comprehensive and accurate resources enriched for transcript-isoform and splice-junction information and computational tools designed to integrate both gene and transcript-level analyses of RNA-sequencing data. Here, an existing and large RNA-sequencing dataset was reanalysed to study seasonal changes in differential gene and transcript expression, alternative splicing, and differential transcript usage in the leaf transcriptome of the perennial evergreen weed Arabidopsis halleri using the recently developed “3D-RNA-Seq” application, a tool purpose-built to increase the accessibility of bioinformatic data analyses and improve the understanding of underexplored, transcript-level phenomena of the eukaryotic transcriptome. Thousands of differentially expressed and spliced genes and isoforms with differential usage were found that were associated with one or more of the four seasonal transitions and were mainly enriched for season-specific stress responses and mRNA processing functions. Transitions into and out of winter exhibited the most transcriptomic changes, and it was also found that most differentially alternatively spliced genes (614/1,013) were not also differentially expressed. These results deeply characterise the seasonal plant transcriptome and indicate alternative splicing as an important and independent mechanism for seasonal adaptation in A. halleri.
AUTHOR SUMMARY
How living organisms adapt to a changing world is a fundamental question of biology. Whereas natural selection makes organisms adapted to their environment by changing the genetic make-up of populations across generations, individual organisms adapt to changes in their immediate environment by altering how their genetic material is used and regulated. Plants make studying this process easy, as all their biology changes in accordance with the seasons. The mainstay of such research has involved measuring which genes are being turned on and off in different environmental contexts and relating their functions to survival, but genes can not only change their expression to meet environmental challenges but also diversify their products by a process called alternative splicing. Here we reanalyse the largest existing gene expression dataset for the well-studied plant Arabidopsis to give one of the most comprehensive descriptions of how alternative splicing changes across seasons.
INTRODUCTION
Understanding how organisms adapt to their environment is a fundamental objective of biology. Whilst genomic tools such as next-generation sequencing (NGS) platforms are regularly used to discern how organisms adapt to their environment over the course of evolution, by enabling nucleotide-resolution analyses of a species’ DNA content (Stapley et al., 2010), most environmental change occurs within an organism’s lifetime and far outpaces the utility of adaptation by natural selection. Adapting to environmental change occurring within an organism’s life requires not varying the features of the genome itself but rather how those features are regulated and expressed in response to ever-changing environmental demands, processes that have come to embody central themes in the “post-genomic” era of biology (McGettigan, 2013; Snyder et al., 2020). The shifting seasons exemplify an environmental change likely to be encountered during an organism’s lifetime, with each season presenting a unique set of biotic and abiotic challenges to survival and reproduction. Plants epitomise seasonal biology, with much of their physiology and lifecycle being shaped by environmental factors like annual fluctuations in temperature and day-length, including the induction of germination (Penfield, 2017), flowering (Searle and Coupland, 2004), and leaf senescence (Guo and Gan, 2005). Accordingly, plants have long served as useful model organisms for fundamental research into the nature of physiological adaptation to changing environments.
Such plant phenological events, in addition to seasonal changes in molecular-level physiological processes like nutrient acquisition (Nord and Lynch, 2009) and photosynthesis (Gu et al., 2003) are ultimately mediated by subcellular molecular machinery controlled by environmentally responsive gene expression networks. Studying seasonally timed changes in plant gene co-expression can therefore enable detailed characterisation of this molecular machinery and further our appreciation for biological adaptation, however such research has tended to occur in controlled laboratory environments (Chaiwanon et al., 2016) or explore the causal role of only one or few selected genes (Aikawa et al., 2010; Satake et al., 2013). NGS of the total RNA in a biological sample (RNA-Seq) provides a high-resolution method for studying the system-level gene expression profile (i.e., “transcriptome”) of plants in different conditions (Abbai et al., 2017) and in their natural habitats (e.g., Champigny et al., 2013; Plessis et al., 2015; Lopez et al., 2019), and can be used to identify functional signatures of adaptation. For example, in a two-year RNA-Seq experiment Nagano et al. (2019) recently captured seasonal dynamics in the leaf transcriptome of a natural population of the perennial evergreen weed A. halleri ssp. gemmifera. In addition to describing consistent patterns in the average monthly transcriptome across each study year, they found 7,185 diurnally and 2,879 seasonally oscillating genes, and used controlled environment experiments wherein they manipulated the natural phase between changing temperature and day length to show that these seasonal transcriptome dynamics were regulated more strongly by temperature fluctuations and have been adapted to improve several phenological measures of fitness.
By producing a detailed characterization of how leaf gene expression responds to the shifting seasons, Nagano et al. (2019) provide important insights into environmental adaptations of the plant transcriptome. However, altering gene expression is only one mechanism that organisms can use to better align their physiology to emerging environmental pressures. Besides differential gene expression (DGE), post-transcriptional regulation at the transcript-level can be used to control the proportion of transcript isoforms expressed by a single gene via differential alternative splicing (DAS) of pre-mRNA molecules (Chen and Manley, 2009). DAS is significantly less understood than DGE but is emerging as an important regulatory mechanism for many adaptive plant processes (e.g., Ding et al., 2014; Thatcher et al., 2017; Calixto et al., 2018). The increased diversity of transcriptional products resulting from DAS creates an additional layer of transcriptomic complexity that is accessible from analyses of RNA-Seq data. For example, single transcript isoforms, similarly to single genes, can be differentially expressed across conditions (differential transcript expression: DTE), or can significantly increase in abundance relative to others (differential transcript usage: DTU).
Complementing gene-level analyses of DGE with transcript-level analyses of DAS, DTE and DTU can radically improve our understanding of the environmental regulation of physiological adaptation. Here we investigate such features of the transcriptome in leaves of Arabidopsis halleri ssp. gemmifera and characterise their activity in seasonal adaptation by reanalysing RNA-Seq data collected by Nagano et al. (2019) using the recently developed 3D-RNA-Seq tool (Guo et al., 2020). An understanding of how transcript-level adaptations of the A. halleri transcriptome help to sustain biological fitness across changing seasonal conditions will not only provide technical details left unexplored by Nagano et al. (2019) but will help to foster a more integrated view of biological adaptation and identify areas for future study.
METHODS
Data Collection and RNA-Sequencing
This study involved the analysis of an existing RNA-Seq dataset of leaf samples taken from a natural population of Arabidopsis halleri ssp. gemmifera over the course of two years. Details of leaf sample collection and RNA-sequencing protocols can be found in the original article (Nagano et al., 2019). Briefly, young (~10 mm) leaves were collected from 6 clonal patches of multiple A. halleri ssp. gemmifera rosettes (one randomly selected leaf per patch) growing naturally at the Omoide River (Japan) every week from 5th July, 2011 to 26th June, 2013, totalling 490 leaf samples across 88 weeks. Leaves were then rapidly treated with RNAlater (Thermo Fisher Scientific) solution before being preserved at -20°C prior to having their RNA extracted and sequenced. Total leaf RNA was extracted using the RNeasy Plant Mini Kit (Quiagen), quantified using a Qubit (Thermo Fisher Scientific) or Quantus (Promega) Fluorometer, and quality-assessed using a Bioanalyzer (Agilent Technologies). 100 or 500 ng of RNA from each sample was used for NGS library preparation (Wang et al., 2011) and reassessed for quality before a maximum of 96 tagged libraries were mixed and sequenced as 50bp single-ended indexed reads over 49 lanes of the HiSeq 2000 (Illumina) sequencer.
Data Pre-Processing, Transcript-Quantification, and Data-Processing
Raw fastq files associated with 490 seasonal experiment samples were acquired from the DDBJ Short Read Archive repository (Accessions: DRA005871, DRA005872, DRA005873, DRA005874, DRA005875 and DRA005876) and quality-assessed with FastQC-0.11.8 (Andrews, 2015) and MultiQC (Ewels et al., 2016). Universal Illumina adapter sequences were removed with the Cutadapt (Martin, 2011) module of TrimGalore-0.6.4 (Krueger, 2015) and samples were reassessed for quality using FastQC and MutliQC.
Transcript-level quantification was run using Salmon-1.2.1 (Patro et al., 2017) in quasi-alignment mode utilising both the --seqBias and --gcBias parameters. The --seqBias flag directs Salmon to learn and correct for random hexamer priming bias that causes some reads to be sequenced preferentially over others due to their starting-sequence motifs, and the --gcBias flag directs Salmon to learn and correct for fragment-level biases in the sequencing of certain fragments due to their GC-content. --gcBias was run because it does not impact quantification accuracy in samples lacking GC bias (only increase run-time), but is demonstrated to reduce systematic quantification errors in samples with significant GC-content sequence bias(es) (Love et al., 2016; Patro et al., 2017), and --seqBias was run because although the two mentioned types of sequencing bias are distinct, they are not entirely independent, and running both flags together directs salmon to run a conditional fragment-GC bias model. Library-type was specified as unstranded (U) or strand-specific reverse (SR) on a sample-dependent basis depending on which had a higher mapping rate.
The AtRTDv2-QUASI Arabidopsis thaliana transcriptome (Zhang et al., 2017) was used as the reference for quasi-alignment due to the fact that no publicly available reference transcriptomes currently exist for A. halleri, but also because AtRTDv2-QUASI is enriched for high-confidence transcript isoform- and splice-junction information for A. thaliana, which is closely related to A. halleri, and thus offers ample opportunities for detecting alternatively spliced genes. AtRTDv2-QUASI was indexed with the --keepDuplicates parameter enabled and a pseudo-kmer length set to 19 bp. The --keepDuplicates flag was enabled because it directs Salmon to disable its default behaviour of removing transcripts with ‘identical’ sequences from the resultant index, which may increase the possibility of highly similar transcript isoforms being inadvertently removed due the heuristics employed within the algorithm for calculating sequence similarity. The quasi-kmer length during indexing was specified as 19 bp, rather than the default of 31 bp (optimised for read fragments ≥75 bp), because for the much shorter ≤49 bp reads used here, it outperformed the default kmer of 31 bp as well as a shorter 26 bp kmer in terms of mapping rates. Following quantification, nine samples were discarded due to having a total of <10^5.5 observed reads, and a further 26 were then discarded due to having ≈0 transcript counts, totalling 455 samples which were progressed onwards for analysis within 3D-RNA-Seq.
Within the 3D-RNA-Seq environment, transcript per million (TPM) quantifications of transcript abundance generated by Salmon were used to estimate transcript- and gene-level pseudo-read counts with TXimport (Soneson et al., 2015) utilising the lengthScaledTPM flag, which corrects count estimations for abundance biases due to library size and read length. Gene- and transcript-level counts per million (CPM) data were then assessed using mean- variance trend plots. The mean-variance trend plots indicated that the data were very highly skewed towards lowly-expressed genes and transcripts. Consistently lowly-expressed genes and transcripts reduce the statistical power of down-stream tests (discussed below) due to increasing the multiple-testing burden on calculations of false discovery rates (FRA) whilst themselves providing little more than statistical noise. Therefore, to negate these limitations and achieve the negative-binomial distribution in the mean-variance trend assumed by downstream statistical analyses, lowly-expressed genes and transcripts (defined here as those that do not have >2CPM in ≥50 samples) were removed. Gene and transcript-level abundances were then normalised with the weighted trimmed mean of M-values (TMM) method, once again correcting for both library- and average fragment-size.
Statistical Analysis: DGE and DTE, DAS, DTU, and Gene Ontology
For statistical discovery of transcriptomic signatures of different seasons weekly gene- and transcript-level log2-CPM data were averaged for each (3 month) season and tested using the Limma-voom Weights pipeline, to reduce the effect of statistical counts outliers across the large dataset. Data were averaged for seasonal months so that an overall ‘signature’ of gene regulation could be identified for each season, and the natural variation across seasonal samples could be collapsed without consideration of meteorological variation. Limma (Ritchie et al., 2015) was used because although it performs similarly to other pipelines (e.g., Sleuth (Pimentel et al., 2017), DESeq2 (Love et al., 2014) and EdgeR (Robinson et al., 2011), it is better able to deal with complex experimental designs and provides more robust transcript-level predictions without reducing statistical stringency (Smyth et al. 2013). Additionally, only Limma and EdgeR allow for integrated analyses at both the gene (DES) and transcript-level (i.e., DAS, DET and DTU), and for the latter EdgeR is known to return far fewer significant results compared to Limma.
To identify changes in gene regulation occurring at seasonal transitions the following four contrast groups were selected: winter versus spring, spring versus summer, summer versus autumn, and autumn versus winter. In each pairwise contrast group differentially expressed genes (DEGs) and transcripts (DETs) were those with an adjusted Benjamini-Hochberg (BH) t-test p value < 0.01 and absolute log2 fold-change (L2FC) > 1. To identify DAS genes, the L2FC of each transcript belonging to a gene was compared to the L2FC for the given gene (i.e., the weighted average of the L2FC of all its transcript isoforms), and all p values were summarised to a single gene-level p value using the F-test method. DAS genes were considered statistically significant when t-test BH-adjusted p < 0.05 and ΔPS (defined as the change in average transcript usage ratios across contrast conditions) > 0.1, where a greater ΔPS is indicative of increased AS events. For DTU testing, the L2FC of each gene transcript isoform was compared against the weighted average L2FC for all the remaining transcript isoforms, and a DTU transcript was considered statistically significant when both L2FC > 1 and t-test BH adjusted p < 0.5.
DE and DAS gene IDs were uploaded to the functional gene annotation website David 6.8 (Dennis et al., 2003) for gene ontology enrichment (GO) analysis. Enriched GO terms categorised as biological process, cellular component, or molecular function were considered significant below a false discovery rate (FDR)-adjusted p value threshold of 0.05 for DGEs or BH adjusted p of 0.05 for DAS genes.
RESULTS
After filtering, 25,292 transcripts encoded by 17,989 genes of the total 82,190 transcripts and 34,212 genes in AtRTDv2-QUASI showed significant levels of expression. A total of 5,571 DEGs were identified, 399 of which were also DAS (DE + DAS) genes. Of the total 12,418 non-DE genes, 614 were DAS (DAS only) genes, meaning that there were 1,013 DAS genes overall (figure 1A). Furthermore, a total of 4,483 DETs were identified, 878 of which were also DTU (DE and DTU) transcripts. Of the 20,809 non-DE transcripts, 945 were DTU (DTU only) transcripts, totalling 1,823 DTU transcripts overall (figure 1B).

The autumn versus winter contrast group showed the greatest change in gene regulation at both the transcript- and gene-level. There were 3,272 DEGs between autumn and winter, 1,279 of which were significantly upregulated and 1,993 of which were downregulated. These DEGs were enriched for biological process GO categories related to general and hormonal responses to abiotic stressors and the regulation of circadian rhythms, cellular components of the chloroplast and molecular functions related to transporter and oxidoreductase activity (figure 2A). There were 481 DAS genes and 2,509 DETs in winter relative to autumn. 1,448 of the DETs were upregulated and 1,061 were downregulated, and 848 transcripts exhibited significant DTU.

Winter versus spring was the contrast group with the second greatest changes in gene regulation. There were 2,935 DEGs in spring compared to winter, 1,636 of which were upregulated and 1,299 of which were downregulated. These DEGs were enriched for biological processes related to general and hormonal responses to abiotic (e.g., salt, cold and desiccation) stressors, photosynthesis and responses to light, and circadian activity, as well as cellular components of the chloroplast and molecular functions associated with chlorophyll- and RNA-binding (figure 2B). 275 genes were DAS in spring relative to winter and there were 2,209 DETs, 1,116 being upregulated, 1,093 being downregulated, and a further 467 DTU transcripts.
There were significantly less changes in gene regulation in the spring versus summer and summer versus autumn contrast groups. There were 1,537 DEGs in summer relative to spring, 579 of which were upregulated and 958 were downregulated. These DEGs were enriched for biological processes associated with immune activity and responses to various (heat, cold, desiccation and high-light intensity) abiotic stressors, and cellular components of the chloroplast (figure 2C). There were 320 DAS genes and 1,079 DETs in summer compared to spring, 468 of which were upregulated and 611 which were downregulated, and a total of 554 DTU transcripts.
These numbers were similar but slightly reduced in autumn compared to summer, where there were 1,005 DEGs, 744 being upregulated and 261 being downregulated. The enriched biological process GO terms were also similar to spring versus summer, being enriched for biological processes related to general immune and heat, cold, light intensity and desiccation responses, and cellular components of the chloroplast, ribosome and cell wall (figure 2D). 261 genes were DAS and 755 transcripts were DE in autumn relative summer, 500 being upregulated, 255 downregulated, and a further 474 were DTU transcripts.
The reduced number of DAS genes compared to DEGs required GO enrichment analysis to be run on the union set of all DAS genes, rather than at individual seasonal transitions. The most significantly enriched biological process for DAS genes across all seasons was the response to cadmium, followed by genes involved in chloroplast organisation and responses to cold- and oxidative-stress (figure 2E).


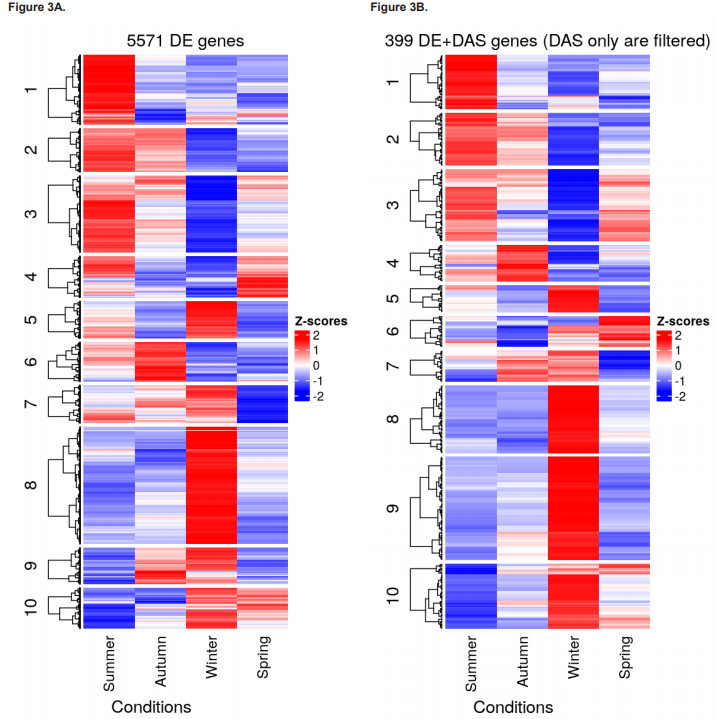
Results of total DEG and DET numbers and their fold-change directions for each contrast group are presented in figures 2F - 2G, and heatmaps showing clustering of DEGs, DE+DAS genes, and DE+DTU transcripts for each season are shown in figures 3A - 3C, respectively. The 10 most significant DEGs, DAS, DETs and DTU results for each pairwise test and their BH-adjusted p value and L2FC are given in table 1.


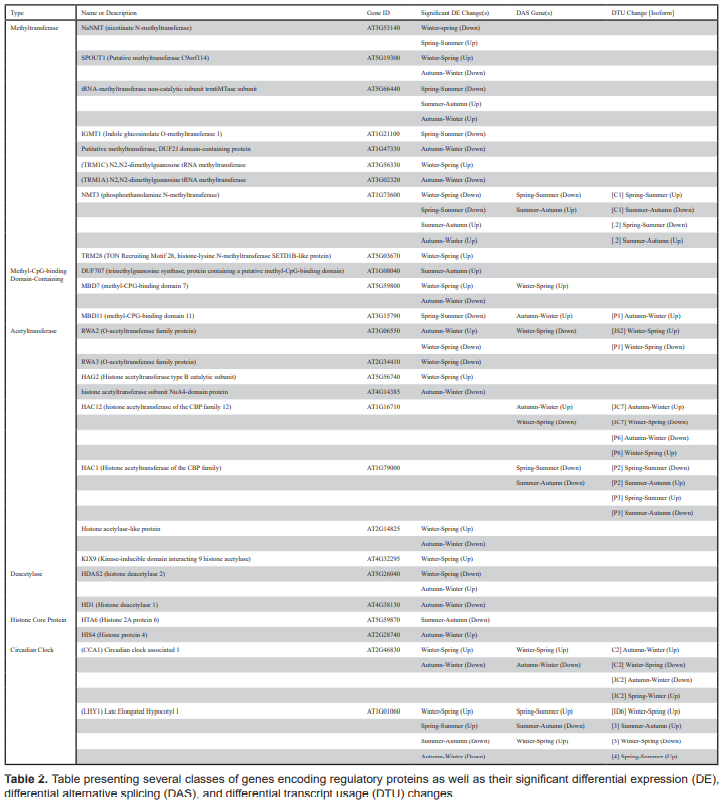
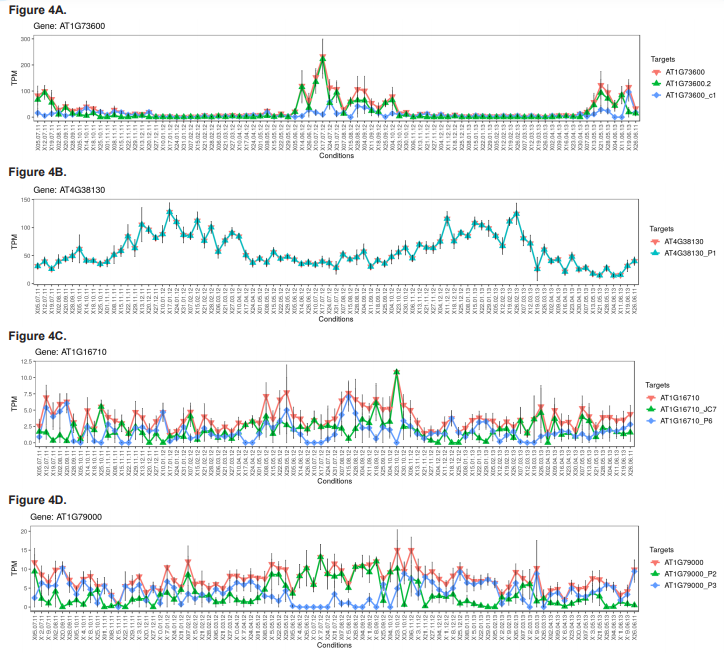
To further explore seasonal regulation of gene expression, a number of differentially regulated genes encoding proteins with functions relating to post-translational protein modifications (methyltransferases, acetyltransferases and deacetyltransferases), protein-DNA interactions (methyl-CpG-binding domains), nucleosome structure (i.e., histone core proteins) and core circadian clock genes were identified and explored. Details of these genes are presented in table 2. The weekly TPM of a select number of these genes are plotted across the three consecutive study years (figure 4) and are discussed below.

DISCUSSION
The objective of this work was to increase the resolution of the seasonal leaf transcriptome dynamics recently uncovered in A. halleri by Nagano et al. (2019) by reanalysing data from their seasonal experiment to include changes in regulation occurring at the transcript-level. Identifying transcript-level changes in gene regulation at seasonal transitions effectively serves to capture novel ‘signatures’ of adaptation to each emerging season that can inform future single-gene or system-level research and help to provide a more holistic perspective of plant environmental physiology. Here, such signatures of adaptation therefore concerned not only DGE but also DTE, DAS and DTU, all of which were significantly increased transitioning in and out of winter relative to transitioning in and out of summer (figures 2F to 2G and figure 3). Of particular note is that most DAS genes (614/1,013) were not also DE, highlighting the seasonal import of DAS in its own right, not only as an aside to DGE.
Whilst seasonal DEGs were largely enriched for biological processes related to various (e.g., cold- and heat-associated) stressors and control of circadian rhythms, DAS genes, besides also exhibiting some enrichment for cold- and heat- (i.e.. oxidative stress) responsive processes, showed a great enrichment for chloroplast-related genes and organisation, with molecular functions additionally linked to protein-binding, GTPase and mRNA-binding activities (figure 2E). In addition to the myriad ways chloroplasts are known to mediate organismal energy homeostasis in changing environments (Demmig-Adams et al., 2014), that chloroplast-related genes and processes are so enriched in the seasonally DAS gene-set is particularly interesting due to the now elucidated role of light-induced regulation of DAS by A. thaliana chloroplasts in laboratory studies (Petrillo et al., 2014; Hertz et al., 2019), particularly because in these studies light-inducible DAS genes were enriched with functions relating to RNA-binding and processing, another enriched seasonally DAS GO term here (figure 2E). This may suggest the chloroplast as a ‘hub’ for the regulation of A. halleri seasonal DAS in nature, and is consistent with the possibility that, at least in the leaves, seasonal DAS may correlate more strongly with fluctuations in light than of temperature. Whilst this would be a significant finding in that it would indicate seasonal regulation of DE and DAS by two separate environmental stimuli, further work would still be needed to clarify if it is seasonally DAS genes per se that are light-responsive, or only some smaller proportion. One potential method to test this would be that employed by Nagano et al. (2019) in their initial study. Namely, to artificially manipulate the period of both light-dark cycles and temperature fluctuations across several months and correlate their effect on transcriptome dynamics, but including DAS, not only DGE. Whilst this reanalysis had been planned for the present work, it was not completed due to time constraints.
The other, and indeed most enriched GO term for seasonally DAS genes was response to cadmium (Cd), a widespread and polluting heavy metal released into the environment by human activities like agriculture, mining, and industry. This result appears anomalous until it is noted that the Omoide-gawa River in Hyogo, where the plants of this study were situated, belongs to the Ichi river basin, which, besides being significantly contaminated with Cd released in part by a nearby mine (Asami et al., 1983) and a source of Cd-associated human “tai-itai” disease (Nogawa et al., 2006), is adjoined to the Jinzu river basin, the single most Cd polluted area in Japan (Aoshima and Horiguchi, 2019). However, why seasonally DAS genes are so functionally enriched for responding to Cd yet this same GO term is not enriched at any seasonal transition within the DEGs is unclear, and is not aided by the lack of study into the effects of Cd on plant DAS, highlighting at once both the value and need for regularly integrating transcript-level analyses into transcriptomic projects.
Several protein-coding genes with known or putative regulatory functions were also investigated as part of this study (table 2). This included genes encoding enzymes that mediate post-translational protein modifications such as methylation, acetylation and deacetylation, as well as CpG-domain-containing proteins, histone proteins and core clock proteins. Histone methyltransferase and acetylase/deacetylase-encoding genes were of particular interest due to their widespread role in the epigenetic regulation of gene expression and potential for widespread seasonal DAS. Whilst most methyltransferase-encoding genes were found to be only DE, including genes encoding a histone-lysine N-methyltransferase (trm28) and four proteins involved in the methylation of either transfer-RNAs (At5g66440, trmc1 and trmca) or microRNAs (c9orf114), nmt3, which encodes a phosphoethanolamine N-methyltransferase (PEAMT) was DE, DAS, and exhibited four DTU events, all of which were associated with entering and leaving summer (figure 4A). PEAMTs are crucial to the production of phosphatidylcholine (PC), the main non-plastid membrane phospholipid in eukaryotes and a central precursor for various lipid-based signalling molecules important to many essential plant cellular processes and stress responses (e.g., Chen et al., 2018). Besides potentially indicating a significant reorganisation of the structure of plant signalling networks during summer, this finding is seemingly the first time a role for DAS or DTU in PC production, an important and highly studied process, has been comprehensively reported.
In addition to methyltransferase enzymes, three genes encoding proteins containing methyl-CpGbinding domains (MBDs) were also explored. Whilst duf707 was only DE, both mbd7 and mbd11 were DAS, with mbd11 also having a significant DTU event moving from autumn to winter. MBDs are the primary ‘reader’ molecules responsible for detecting and binding DNA sequences of methylated CpG dinucleotides and are therefore crucial to the specificity of DNA-protein interactions underlying the epigenetic repression of gene expression (Du et al., 2015). Their varying DE and DAS (mbd7 having greater expression and AS in spring and mbd11 in winter) is therefore a likely indication of their different roles in the upstream repression of different seasonally relevant gene networks. Whilst regulation of DAS by CBD proteins is well-known (e.g., Young et al., 2005), regulation of CBD protein function by DAS has received less attention, but here provides a parsimonious explanation as to how some seasonal changes in DGE are likely to be regulated. An interesting future question is what proportion of seasonal DEGs are repressed by such upstream DAS of CBD genes.
Interestingly, whilst many acetyltransferase and histone acetyltransferase-encoding genes, as well as genes encoding histone proteins and histone deacetylases (figure 4B) were found to be DE-only, two histone acetyltransferase-encoding genes, hac1 and hac12, exhibited extensive seasonal DAS and DTU events without any change in their expression (figures 4C and 4D). Unlike MBD-encoding genes, regulation of histone acetyltransferase function by DAS is relatively well documented (e.g., Lois et al., 2007). However, whilst the diversification of plant histone acetyltransferase proteins by DAS is well studied in the context of evolutionary diversification (e.g., Pandey et al., 2002), its role in seasonal adaptation is less understood, but likely to yield novel mechanistic insights into plant environmental physiology.
We also explored two Arabidopsis clock genes, lhy1 and cca1 (figures 4E and 4F). Both of these genes encode myb transcription factors involved in a negative feedback loop closely associated with the Arabidopsis circadian oscillator system (Carré and Kim, 2002), which times the expression of one third of the genes in the Arabidopsis genome (Covington et al., 2008). Due to the central role of the Arabidopsis clock system in timing many seasonal physiological traits like germination and flowering, lhy1 and cca1 are both intensely studied and known to undergo extensive DAS, particularly in response to changing temperatures (Filichkin and Mockler, 2012; James et al., 2012; Park et al., 2012; Capovilla et al., 2015). These findings are reinforced here. Besides both being generally downregulated in autumn and winter and upregulated in spring and summer, lhy1 and cca1 together showed the most extensive DAS and DTU events of all of the explored regulatory genes, with cca1 even being in the top 10 overall DAS genes for winter-spring (table 1). This study therefore provides comprehensive and naturalistic support for temperature dependent AS of lhy1 and cca1 as a means for linking Arabidopsis circadian rhythms to yearly temperature changes.
The present study provides a detailed characterisation of the A. halleri transcriptome in response to natural seasonal changes at the gene- and transcript levels that can be used and further explored by others researching seasonal plant phenotypes and adaptation. It is however important to be mindful that to properly understand how the results presented here relate to plant seasonal adaptation, studies utilising proteomic, epigenomic and metabolomic methods should be employed to elucidate how changes in the transcriptome translate to changes in phenotypes, with the ultimate goal to causally link these phenotypes to demonstrations of adaptive changes in fitness outcomes. It is also important to be aware of the limitations and potential confounders, the primary here being the somewhat unusually low gene and transcript count numbers, which undoubtedly increase the inherent statistical noise in the data and chance of spurious results and conclusions. Likely explanations for the generally very low counts (and the especially low ≈0 counts of 26 excluded samples) are a combination of a high proportion of small library sizes, generally low (63±10%) mapping rates, and some degree of inaccurate strand information. Besides the reasons already discussed (§2.2), a further explanation for these observed low-counts data arises from the fact that an A. thaliana reference was used to study the A. halleri transcriptome. Although not the first time this had been done, and somewhat unavoidable due to the lack of publicly available A. halleri reference transcriptome, this outcome potentially indicates greater genomic differences between these two species than is often assumed, and serves as a reminder of the increasing value of dedicated efforts to build accessible and accurate bioinformatics resources. Moreover, it underlines the need for diversifying the base of model organisms within biology, as understanding biological phenomena ultimately demands accounting for their natural biodiversity.
ACKNOWLEDGMENTS
The authors would like to thank the University of Dundee Summer School for funding the work presented in this article. DP would like to thank The University of Dundee for providing the funding that allowed this project to be carried out, The James Hutton Institute in Dundee for allowing access to their high-performance computer cluster, and RZ and WG for their generous support and supervision.
REFERENCES
Abbai, R., Subramaniyam, S., Mathiyalagan, R. and Yang, D.C. (2017). Functional genomic approaches in plant research. In Plant Bioinformatics (215-239). https://doi.org/10.1007/978-3-319-67156-7_8
Aikawa, S., Kobayashi, M.J., Satake, A., Shimizu, K.K. and Kudoh, H. (2010). Robust control of the seasonal expression of the Arabidopsis FLC gene in a fluctuating environment. Proceedings of the National Academy of Sciences, 107(25), 11632-11637. https://doi.org/10.1073/pnas.0914293107
Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data.
Aoshima, K. and Horiguchi, H. (2019). Historical Lessons on Cadmium Environmental Pollution Problems in Japan and Current Cadmium Exposure Situation. In Cadmium Toxicity (3-19). https://doi.org/10.1007/978-981-13-3630-0_1
Asami, T., Kubota, M., Homma, S., Wada, T. and Nakashima, K. (1983). Pollution of unpolished rice produced in the Ichi and Maruyama river basins by cadmium discharged from Ikuno mine, Hyogo prefecture [J.]. Japanese Journal of Soil Science and Plant Nutrition.
Calixto, C.P., Guo, W., James, A.B., Tzioutziou, N.A., Entizne, J.C., Panter, P.E., Knight, H., Nimmo, H.G., Zhang, R. and Brown, J.W. (2018). Rapid and dynamic alternative splicing impacts the Arabidopsis cold response transcriptome. The Plant Cell, 30(7), 1424-1444. https://doi.org/10.1105/tpc.18.00177
Capovilla, G., Pajoro, A., Immink, R.G. and Schmid, M. (2015). Role of alternative pre-mRNA splicing in temperature signaling. Current opinion in plant biology, 27, 97-103. https://doi.org/10.1016/j.pbi.2015.06.016
Carré, I.A. and Kim, J.Y. (2002). MYB transcription factors in the Arabidopsis circadian clock. Journal of experimental botany, 53(374), 1551-1557. https://doi.org/10.1093/jxb/erf027
Chaiwanon, J., Wang, W., Zhu, J.Y., Oh, E. and Wang, Z.Y. (2016). Information integration and communication in plant growth regulation. Cell, 164(6), 1257-1268. https://doi.org/10.1016/j.cell.2016.01.044
Champigny, M.J., Sung, W.W., Catana, V., Salwan, R., Summers, P.S., Dudley, S.A., Provart, N.J., Cameron, R.K., Golding, G.B. and Weretilnyk, E.A. (2013). RNA-Seq effectively monitors gene expression in Eutrema salsugineum plants growing in an extreme natural habitat and in controlled growth cabinet conditions. BMC Genomics, 14(1), 578. https://doi.org/10.1186/1471-2164-14-578
Chen, M. and Manley, J.L. (2009). Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nature reviews Molecular cell biology, 10(11), 741-754. https://doi.org/10.1038/nrm2777
Chen, W., Salari, H., Taylor, M.C., Jost, R., Berkowitz, O., Barrow, R., Qiu, D., Branco, R. and Masle, J. (2018). NMT1 and NMT3 N-methyltransferase activity is critical to lipid homeostasis, morphogenesis, and reproduction. Plant physiology, 177(4), 1605-1628. https://doi.org/10.1104/pp.18.00457
Covington, M.F., Maloof, J.N., Straume, M., Kay, S.A. and Harmer, S.L. (2008). Global transcriptome analysis reveals circadian regulation of key pathways in plant growth and development. Genome biology, 9(8), p.R130. https://doi.org/10.1186/gb-2008-9-8-r130
Demmig-Adams, B., Stewart, J.J. and Adams III, W.W. (2014). Multiple feedbacks between chloroplast and whole plant in the context of plant adaptation and acclimation to the environment. Philosophical Transactions of the Royal Society B: Biological Sciences, 369(1640), 20130244. https://doi.org/10.1098/rstb.2013.0244
Dennis, G., Sherman, B.T., Hosack, D.A., Yang, J., Gao, W., Lane, H.C. and Lempicki, R.A. (2003). DAVID: database for annotation, visualization, and integrated discovery. Genome biology, 4(9), 1-11. https://doi.org/10.1186/gb-2003-4-9-r60 Available at: https://david.ncifcrf.gov/
Ding, F., Cui, P., Wang, Z., Zhang, S., Ali, S. and Xiong, L. (2014). Genome-wide analysis of alternative splicing of pre-mRNA under salt stress in Arabidopsis. BMC genomics, 15(1), 431. https://doi.org/10.1186/1471-2164-15-431
Du, Q., Luu, P.L., Stirzaker, C. and Clark, S.J. (2015). Methyl-CpG-binding domain proteins: readers of the epigenome. Epigenomics, 7(6), 1051-1073. https://doi.org/10.2217/epi.15.39
Ewels, P., Magnusson, M., Lundin, S. and Käller, M. (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 32(19), 3047-3048. https://doi.org/10.1093/bioinformatics/btw354
Filichkin, S.A. and Mockler, T.C. (2012). Unproductive alternative splicing and nonsense mRNAs: a widespread phenomenon among plant circadian clock genes. Biology direct, 7(1), 20. https://doi.org/10.1186/1745-6150-7-20
Gu, L., Post, W.M., Baldocchi, D., Black, T.A., Verma, S.B., Vesala, T. and Wofsy, S.C. (2003). Phenology of vegetation photosynthesis. In Phenology: An integrative environmental science. 467-485. Doi: 10.1007/978-94-007-0632-3_29
Guo, W., Tzioutziou, N.A., Stephen, G., Milne, I., Calixto, C.P., Waugh, R., Brown, J.W. and Zhang, R. (2020). 3D RNA-seq: A powerful and flexible tool for rapid and accurate differential expression and alternative splicing analysis of RNA-seq data for biologists. RNA biology, 1-14. https://doi.org/10.1080/15476286.2020.1858253
Guo, Y. and Gan, S. (2005). Leaf senescence: signals, execution, and regulation. Current topics in developmental biology, 71, 83-112. https://doi.org/10.1016/S0070-2153(05)71003-6
Herz, M.A.G., Kubaczka, M.G., Brzyżek, G., Servi, L., Krzyszton, M., Simpson, C., Brown, J., Swiezewski, S., Petrillo, E. and Kornblihtt, A.R. (2019). Light regulates plant alternative splicing through the control of transcriptional elongation. Molecular cell, 73(5), 1066-1074. https://doi.org/10.1016/j.molcel.2018.12.005
James, A.B., Syed, N.H., Bordage, S., Marshall, J., Nimmo, G.A., Jenkins, G.I., Herzyk, P., Brown, J.W. and Nimmo, H.G. (2012). Alternative splicing mediates responses of the Arabidopsis circadian clock to temperature changes. The Plant Cell, 24(3), pp.961-981. https://doi.org/10.1105/tpc.111.093948
Krueger, F. (2015). Trim galore. A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files, 516, p.517.
Lois, S., Blanco, N., Martínez-Balbás, M. and de la Cruz, X. (2007). The functional modulation of epigenetic regulators by alternative splicing. BMC genomics, 8(1), 252. https://doi.org/10.1186/1471-2164-8-252
Lopez, L., Bellis, E.S., Wafula, E., Hearne, S.J., Honaas, L., Ralph, P.E., Timko, M.P., Unachukwu, N., DePamphilis, C.W. and Lasky, J.R. (2019). Transcriptomics of host-specific interactions in natural populations of the parasitic plant purple witchweed (Striga hermonthica). Weed Science, 67(4), 397-411. doi:10.1017/wsc.2019.20
Love, M.I., Hogenesch, J.B. and Irizarry, R.A. (2016). Modeling of RNA-seq fragment sequence bias reduces systematic errors in transcript abundance estimation. Nature biotechnology, 34(12), p.1287. https://doi.org/10.1038/nbt.3682
Love, M.I., Huber, W. and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology, 15(12), 550. https://doi.org/10.1186/s13059-014-0550-8
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal, 17(1), 10-12. https://doi.org/10.14806/ej.17.1.200
McGettigan, P.A. (2013). Transcriptomics in the RNA-seq era. Current opinion in chemical biology, 17(1), 4-11. https://doi.org/10.1016/j.cbpa.2012.12.008
Nagano, A.J., Kawagoe, T., Sugisaka, J., Honjo, M.N., Iwayama, K. and Kudoh, H. (2019). Annual transcriptome dynamics in natural environments reveals plant seasonal adaptation. Nature plants, 5(1), 74-83. https://doi.org/10.1038/s41477-018-0338-z
Nogawa, K. (2006). Tissue cadmium (Cd) concentrations of people living in a Cd polluted area, Japan. Biometals, 19(5), 521-525. DOI 10.1007/s10534-005-5619-0
Nord, E.A. and Lynch, J.P. (2009). Plant phenology: a critical controller of soil resource acquisition. Journal of experimental botany, 60(7), 1927-1937. https://doi.org/10.1093/jxb/erp018
Pandey, R., MuÈller, A., Napoli, C.A., Selinger, D.A., Pikaard, C.S., Richards, E.J., Bender, J., Mount, D.W. and Jorgensen, R.A. (2002). Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes. Nucleic acids research, 30(23), 5036-5055. https://doi.org/10.1093/nar/gkf660
Park, M.J., Seo, P.J. and Park, C.M. (2012). CCA1 alternative splicing as a way of linking the circadian clock to temperature response in Arabidopsis. Plant signaling & behavior, 7(9), 1194-1196. https://doi.org/10.4161/psb.21300
Patro, R., Duggal, G., Love, M.I., Irizarry, R.A. and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nature methods, 14(4), 417-419. https://doi.org/10.1038/nmeth.4197
Penfield, S. (2017). Seed dormancy and germination. Current Biology, 27(17), 874-878. https://doi.org/10.1016/j.cub.2017.05.050
Petrillo, E., Herz, M.A.G., Fuchs, A., Reifer, D., Fuller, J., Yanovsky, M.J., Simpson, C., Brown, J.W., Barta, A., Kalyna, M. and Kornblihtt, A.R. (2014). A chloroplast retrograde signal regulates nuclear alternative splicing. Science, 344(6182), 427-430. DOI: 10.1126/science.1250322
Pimentel, H., Bray, N.L., Puente, S., Melsted, P. and Pachter, L. (2017). Differential analysis of RNA-seq incorporating quantification uncertainty. Nature methods, 14(7), 687. https://doi.org/10.1038/nmeth.4324
Plessis, A., Hafemeister, C., Wilkins, O., Gonzaga, Z.J., Meyer, R.S., Pires, I., Müller, C., Septiningsih, E.M., Bonneau, R. and Purugganan, M. (2015). Multiple abiotic stimuli are integrated in the regulation of rice gene expression under field conditions. Elife, 4, .e08411. Doi: 10.7554/eLife.08411
Ritchie, M.E., Phipson, B., Wu, D.I., Hu, Y., Law, C.W., Shi, W. and Smyth, G.K. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research, 43(7), e47-e47. https://doi.org/10.1093/nar/gkv007
Robinson, M.D., McCarthy, D.J. and Smyth, G.K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26(1), 139-140. https://doi.org/10.1093/bioinformatics/btp616
Satake, A., Kawagoe, T., Saburi, Y., Chiba, Y., Sakurai, G. and Kudoh, H. (2013). Forecasting flowering phenology under climate warming by modelling the regulatory dynamics of flowering-time genes. Nature communications, 4(1), 1-8. https://doi.org/10.1038/ncomms3303
Searle, I. and Coupland, G. (2004). Induction of flowering by seasonal changes in photoperiod. The EMBO Journal, 23(6), 1217-1222. https://doi.org/10.1038/sj.emboj.7600117
Snyder, M.P., Gingeras, T.R., Moore, J.E., Weng, Z., Gerstein, M.B., Ren, B., Hardison, R.C., Stamatoyannopoulos, J.A., Graveley, B.R., Feingold, E.A. and Pazin, M.J. (2020). Perspectives on ENCODE. Nature, 583(7818), 693-698. https://doi.org/10.1038/s41586-020-2449-8
Soneson, C., Love, M.I. and Robinson, M.D. (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research, 4. doi: 10.12688/f1000research.7563.2
Stapley, J., Reger, J., Feulner, P.G., Smadja, C., Galindo, J., Ekblom, R., Bennison, C., Ball, A.D., Beckerman, A.P. and Slate, J. (2010). Adaptation genomics: the next generation. Trends in ecology and evolution, 25(12), 705-712. https://doi.org/10.1016/j.tree.2010.09.002
Thatcher, S.R., Danilevskaya, O.N., Meng, X., Beatty, M., Zastrow-Hayes, G., Harris, C., Van Allen, B., Habben, J. and Li, B. (2016). Genome-wide analysis of alternative splicing during development and drought stress in maize. Plant physiology, 170(1), 586-599. https://doi.org/10.1104/pp.15.01267
Voelckel, C., Gruenheit, N. and Lockhart, P. (2017). Evolutionary transcriptomics and proteomics: insight into plant adaptation. Trends in plant science, 22(6), 462-471. https://doi.org/10.1016/j.tplants.2017.03.001
Wang, L., Si, Y., Dedow, L.K., Shao, Y., Liu, P. and Brutnell, T.P. (2011). A low-cost library construction protocol and data analysis pipeline for Illumina-based strand-specific multiplex RNA-seq. PloS one, 6(10), e26426. https://doi.org/10.1371/journal.pone.0026426
Young, J.I., Hong, E.P., Castle, J.C., Crespo-Barreto, J., Bowman, A.B., Rose, M.F., Kang, D., Richman, R., Johnson, J.M., Berget, S. and Zoghbi, H.Y. (2005). Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proceedings of the National Academy of Sciences, 102(49), 17551-17558. https://doi.org/10.1073/pnas.0507856102
Zhang, R., Calixto, C.P., Marquez, Y., Venhuizen, P., Tzioutziou, N.A., Guo, W., Spensley, M., Entizne, J.C., Lewandowska, D., ten Have, S. and Frei dit Frey, N. (2017). A high quality Arabidopsis transcriptome for accurate transcript-level analysis of alternative splicing. Nucleic acids research, 45(9), 5061-5073. https://doi.org/10.1093/nar/gkx267